
The Gene Therapy Pipeline — And The Biggest Challenges Facing Developers
By Claudia Dall’Osso, Ph.D., and Akash Saini, Ph.D., Decision Resources Group (DRG)

Gene therapy has moved from concept to reality over a decades-long journey during which the field has hurdled technological and clinical challenges. From the advent of enabling viral vectors for effective gene delivery, through the emergence of safety signals (e.g., immune, malignancy) in pilot trials, methodological refinements have yielded better and safer methods of gene manipulation and driven a flurry of R&D.
With several first-in-class gene therapies now approved, distinct new challenges — particularly surrounding evidence generation and market access — are now coming into focus. In this two-part article, we first provide a high-level overview of the gene therapy pipeline for non-oncology rare diseases and discuss key issues impacting the clinical development of these emerging therapeutics. In Part 2, we discuss value assessment challenges affecting the commercialization of gene therapies, review the path to market for Spark Therapeutics’ trailblazing gene therapy Luxturna, and summarize key considerations for gene therapy developers striving to bring innovative therapeutics to market.
The Gene Therapy Pipeline, By The Numbers
Generally speaking, there are three main types of gene manipulation strategies with current or future clinical applications. Gene replacement exogenously delivers a functional copy of a gene to restore function lost due to a mutation (e.g., Luxturna, which delivers a functional copy of RPE65 to photoreceptors). Antisense oligonucleotides or small interfering RNAs silence gene expression at the level of translation (e.g., Exondys 51 for Duchenne muscular dystrophy and Spinraza for spinal muscular atrophy). Gene editing is designed to correct a mutation in a dysfunctional gene in situ through the use of engineered nucleases (e.g., zinc finger nucleases [ZFN] technology) or Cas9 nucleases (i.e., CRISPR/Cas9 technology).
There are nearly 150 gene therapies currently in development (Figure 1). Approximately 80 percent of rare diseases are thought to be monogenic in nature1and, thus, amenable to interventions targeting a single gene. Unsurprisingly, nearly 70 percent of emerging gene therapies are being tested as a rare disease treatment: metabolic, ophthalmological, and hematological rare diseases together comprise more than half of the disease targets in the gene therapy pipeline. Indeed, the accessibility and immune-privileged status of the eye have rendered ophthalmic diseases attractive early targets for gene therapy developers; many mid- or late-stage programs — including the first approved gene therapy in the United States — are designed to treat eye diseases.
Gene replacement strategies account for more than two-thirds of the gene therapy pipeline, and nearly 30 percent of emerging programs are RNAi or antisense oligonucleotides. Gene editing programs comprise only a very small fraction of the gene therapy pipeline, and all are in early stages of development. Several ZFN-based programs from Sangamo Therapeutics are in Phase 1/2 trials for the treatment of mucopolysaccharidosis I and II, and hemophilia A and B. Although top-of-mind for many in the industry and supported by a blistering pace of technological advances, CRISPR/Cas9 programs remain in the discovery phase of development. Nonetheless, the technology has yielded impressive preclinical data in multiple genetic and infectious disease models, including Duchenne muscular dystrophy, type-1 tyrosinemia and hepatitis C virus.
At least 10 companies in the U.S. have CRISPR-based programs in their preclinical/discovery pipeline. Although challenges pertaining to the specificity, efficiency, and delivery of gene-editing components remain, experts expect the technology will evolve faster than conventional gene therapy and offer greater potential for a permanent cure given the in situ nature of the correction. We expect at least two companies to move toward human trials of CRISPR-based medications in 2018; Swiss biotech CRISPR Therapeutics, along with partner Vertex, is poised to submit an investigational new drug (IND) application for a sickle cell disease therapy, and EDITAS medicine, based in Cambridge, Massachusetts, is expected to file an IND for a program targeting Leber Congenital Amaurosis (LCA) 10.
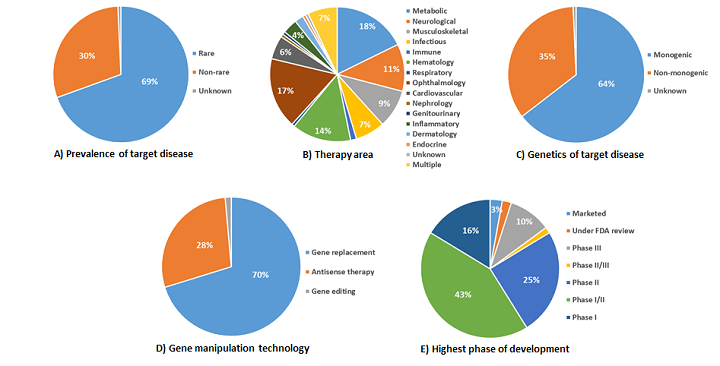
Figure 1: The gene therapy pipeline by the numbers. Here, we segment the gene therapy pipeline by: A) prevalence of target disease, B) therapy area, C) genetics of target disease, D) highest phase of development of the therapy, and E) gene manipulation technology.
Clinical Developmental Challenges Facing Gene Therapies In Rare Diseases
Clinical trial design: Randomized, placebo-controlled clinical trials represent the gold standard supporting applications for regulatory approval. However, for practical and ethical reasons, such trials are seldom implemented in the rare disease space, and they are especially challenging for gene therapies in ultra-rare monogenic diseases.
The first and most obvious limitation is population size; it is difficult to find and recruit a pool of eligible patients sufficient to conduct robust statistical analyses. For gene therapies offering substantial clinical benefit, a limited data set may suffice. However, when more modest benefits in clinical outcomes are expected/realized, data from a small clinical trial may not unequivocally support the efficacy of a gene therapy, adding a level of uncertainty in the interpretation of the data. And uncertainty breeds suboptimal buy-in from key stakeholders, including regulators, treating physicians, and payers (e.g., Sarepta Therapeutics’ Exondys 51).
Depending on the target tissue affected, the invasive delivery of a gene therapy may be difficult, uncomfortable, or even unethical to sham-control and blind. For instance, the first gene therapy approved (and ultimately withdrawn) in Europe—uniQure/Chiesi’s Glybera—required up to 60 intramuscular injections into the legs. Spark’s Luxturna requires a subretinal surgical procedure. Neither pivotal program included a blinded sham control arm, although Spark separately collected natural history data to support their regulatory application, in alignment with FDA guidance on drug development for rare diseases. In the case of Glybera, no natural history data was presented; although the agent was ultimately approved, only one patient received it post-approval. Clinicians we’ve interviewed reported skepticism about Glybera’s efficacy, lamenting the lack of a control arm to confirm the agent had a meaningful impact on clinical outcomes.
Although natural history data can obviate the need for blinding or a placebo arm, there are limitations to this strategy. A simple change in conditions, such as the use of a background or symptomatic therapy that was not available to historical controls, can confound the resulting data. Furthermore, if patients in the clinical trial carry a specific genetic defect, it is important to evaluate whether the disease severity and progression is comparable with that of the general population. Ultimately, important prognostic factors may not be identified or included in historical data; as such, the FDA guides rare disease developers that historical data suffices only when a treatment’s effect size is large relative to natural variation in a disease course.
Outcome metrics: Developers advancing gene therapies for monogenic rare diseases may need to trail blaze and develop an outcome metric that will appease regulators, payers, patients, and physicians. It is necessary to convince key stakeholders not only of the validity of the metric itself, but also of its correlation to clinically meaningful outcomes. Patient-reported outcomes that measure patients’ daily living activities and quality of life can be particularly powerful in demonstrating to stakeholders that even small efficacy gains can have a meaningful positive impact on patients suffering from progressive, debilitating diseases.
Outcome metrics must be as objective as possible, especially if a control arm is lacking. For example, a typical outcome metric used to capture aerobic capacity and endurance is the six-minute walk test, which measures the distance a patient can walk in six minutes. However, because performance can be colored by a patient’s belief in the therapeutic intervention, this measure is not sufficiently objective for an open-label trial.
To obviate some of these challenges, biomarkers (e.g., production of a transgenic protein) can be used as surrogate endpoints, with the obvious caveat that, while this data can prove the gene therapy is accomplishing its intended goal (i.e., boost production of a given protein), it is often unclear how much protein is fully functional and how much functional protein is needed to deliver a clinically meaningful outcome.
Lingering safety concerns: A recently published article by prominent gene therapy pioneer Dr. James Wilson of the University of Pennsylvania emphasizes the need to carefully consider the dose and delivery of the viral vector that is necessary to reach the target organ; high intravenous doses of adeno-associated viruses led to acute liver failure in non-human primates and proprioceptive deficits in piglets.2 These actions may relate to the ability of a virus to trigger an innate immune response. Although high intravenous doses are not necessary for accessible targets (e.g., the eye), some target diseases require systemic vector delivery — and, thus, may pose a more challenging risk/benefit assessment for the gene therapy approach and may demand more extensive preclinical testing to support the safety of the vector.
In Part 2 of this two-part article, we discuss Spark Therapeutics’ strategy to overcome these clinical development challenges and attain a landmark FDA approval for Luxturna, a gene therapy approved to treat RPE65-mediated retinal dystrophy. We then discuss gene therapy value assessment challenges and cover early observations from the evolving cost-effectiveness discussion surrounding Luxturna.
References:
- Puiu M, et al. Rare diseases, from European resolutions and recommendations to actual measures and strategies. Maedica (Buchar). 2010;5(2):128-131.
- Hinderer C, et al. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum Gene Ther. 2018.
About The Authors:
 Claudia Dall’Osso, Ph.D., is a principal business insights analyst on the Infectious, Niche, and Rare Diseases team at Decision Resources Group, specializing in niche and rare indications. Before joining DRG, she held a management and strategy consultant position at Precision Medicine Group, where she worked for clients in the biopharmaceutical, medical, device and diagnostic industries. Dall’Osso completed her master’s in management at Harvard University; she also holds a Ph.D. in medical genetics from Brescia University in Italy and a B.S./M.S. degree in medical biotechnology from the University of Milano in Italy. You can reach her at cdallosso@teamdrg.com or connect with her on LinkedIn.
Claudia Dall’Osso, Ph.D., is a principal business insights analyst on the Infectious, Niche, and Rare Diseases team at Decision Resources Group, specializing in niche and rare indications. Before joining DRG, she held a management and strategy consultant position at Precision Medicine Group, where she worked for clients in the biopharmaceutical, medical, device and diagnostic industries. Dall’Osso completed her master’s in management at Harvard University; she also holds a Ph.D. in medical genetics from Brescia University in Italy and a B.S./M.S. degree in medical biotechnology from the University of Milano in Italy. You can reach her at cdallosso@teamdrg.com or connect with her on LinkedIn.
 Akash Saini, Ph.D., is a lead analyst with the Infectious, Niche, and Rare Diseases team at Decision Resources Group, where he specializes in a diverse group of rare diseases. He received his Ph.D. in biochemistry and biotechnology from the International Centre for Genetic Engineering and Biotechnology (ICGEB) New Delhi in India and his M.Sc. in biotechnology from Jawaharlal Nehru University, also in India. Prior to joining Decision Resources Group, Saini was a postdoctoral fellow at the University of Massachusetts Medical School, where he studied mitochondrial dysfunction in amyotrophic lateral sclerosis (ALS). You can reach him at asaini@teamdrg.com or connect with him on LinkedIn.
Akash Saini, Ph.D., is a lead analyst with the Infectious, Niche, and Rare Diseases team at Decision Resources Group, where he specializes in a diverse group of rare diseases. He received his Ph.D. in biochemistry and biotechnology from the International Centre for Genetic Engineering and Biotechnology (ICGEB) New Delhi in India and his M.Sc. in biotechnology from Jawaharlal Nehru University, also in India. Prior to joining Decision Resources Group, Saini was a postdoctoral fellow at the University of Massachusetts Medical School, where he studied mitochondrial dysfunction in amyotrophic lateral sclerosis (ALS). You can reach him at asaini@teamdrg.com or connect with him on LinkedIn.