
The Impact Of Regulatory & Analytics Evolution On The Biopharma Industry
By Ajay Babu Pazhayattil, Marzena Ingram, and Naheed Sayeed-Desta

The biopharmaceutical industry is currently going through a period of tremendous change. Regulators, service providers, industry, and patients are increasingly relying on knowledge and intelligence. With the growth of the information industry propagating a knowledge-based global society, the healthcare sector is joining the trend of utilizing the existing body of knowledge to its highest potential. Further, the FDA is promoting efforts to modernize approaches to fulfilling its mandate of protecting and promoting public health. The agency intends to continue applying sound science while approving medicinal products in an efficient and predictable manner, for which data is pivotal. The mandate of the 21st Century Cures Act,1 signed into law on Dec. 13, 2016, is to help accelerate medicinal product development and bring new innovations and advances to patients. The law makes it imperative to accelerate adoption of newer technologies throughout the biopharmaceutical industry to bring lifesaving drugs to patients more quickly. The technology innovations discussed in this article could result in significant adjustments to the way biopharmaceutical businesses operate, hence they are disruptive in nature.
This two-part article speaks to the use of various tools and the emergence of intelligent solutions in the industry. The first part provides an overview of how technological advancements and data analytics are critical in enabling regulators and the life sciences industry. The second part further elucidates the emerging collaborative environment and the future prospect of artificial intelligence (AI) maturing into cross sector intelligence (CSI), a cross sector convergence, generating intelligent decisions based on data sets from multiple sectors for the benefit of patients.
Regulatory Agencies Encourage Use Of New Technologies, Approaches
Health agencies mandate that essential drug products are available to patients in a timely manner. Agencies want to empower patients and provide them with up-to-date information. The FDA has therefore initiated a Patient Engagement Collaborative (PEC), along with a Clinical Trials Transformation Initiative (CTTI), to achieve this empowerment.2 The goal of the PEC is to provide diverse perspectives on patient engagement, transparency, and communication. In addition, the PEC will provide a platform for considering new strategies to enhance patient engagement at the FDA while proposing novel models of collaboration with respect to product development and the FDA review process. The FDA recognizes that knowledge from real-world evidence, derived without randomized clinical trials, has the potential to allow researchers to answer questions about efficacy. This will save time and money while fielding answers relevant to broader patient populations who have limited access to clinical environments. The FDA’s acceptance of a new drug application (NDA) for a novel clinical protocol for lisinopril using crowdsourced data from MS researchers, physicians, and patients is a good example of a first step toward the use of real-world data.3 The plan minimizes the need for site visits while relying on telemonitoring and virtual interaction with the subjects.
FDA Initiatives
As apparent from the Food and Drug Administration Safety and Innovation Act (FDASIA) initiatives, as well as the FDA’s workshop on leveraging quantitative methods and modeling to modernize generic drug development review, held in October 2017, the FDA is committed to promoting modeling and simulation in drug development. In a July 2016 status report to Congress on the Best Pharmaceuticals for Children Act and Pediatric Research Equity Act,4 the FDA discusses the establishment of working groups with FDA participants to identify non-clinical models and use the information to categorize safe and effective starting doses for studies on neonates. The proven scientific approach of modeling and simulation aids in critical drug development decisions such as dosing, drug-drug interaction, and deciding the relevant patient surrogate endpoint. With the growth of large-molecule biopharma candidates and the emergence of biosimilars, the variability and development costs are high; therefore, it becomes imperative to use modeling and simulation (M&S) to improve efficiencies and ensure cost-effectiveness.
In line with ICH Q85 considerations, accepted by the FDA and other major regulatory bodies, quality by design (QbD) concepts for formulation and process development have been used by the biopharma/pharmaceutical industries for several years. Developing a robust control strategy requires incorporation of prior knowledge, applying results of design of experiments (DoE) studies, as well as using risk management and knowledge management methodologies. Incorporating prior knowledge and novel approaches based on newer outcomes calls for accessible and versatile knowledge management solutions. Continued or ongoing process verification (CPV)6 provides assurance during routine production that the process remains in a state of control. Being a regulatory (FDA, EMA, PIC/S) guidance recommendation, the data collection and statistical assessment provides opportunities for continuous improvement activities prior to reaching failure mode. Regulators recommend that manufacturers use quantitative statistical methods whenever appropriate and feasible. Scrutiny of intra- and inter-batch variation is part of a comprehensive continued process verification program under Code of Federal Regulations (CFR) Title 21. Continued process verification is the third process stage after design and qualification. The FDA intends to publish a list of organizations that voluntarily report quality metrics to drive industry participation in regularly reporting quality metrics. This calls for adequate monitoring and reporting tools to meet the regulatory expectations.
The FDA has been at the forefront of modernizing the way drugs are made, including introducing the Emerging Technology Program. One of the biggest initiatives the agency is promoting is transitioning into continuous manufacturing (CM). The U.S. Congress has recognized the potential benefits of CM7 and has authorized grants to support CM studies in drugs and biological manufacturing. The FDA’s post-marketing safety reporting regulations for human drugs and biological products, including blood and blood products, are being revised to better align with International Council on Harmonisation (ICH) guidelines. An update to the reporting requirements is needed in light of current pharmacovigilance practices and safety information sources and to enhance the quality of safety reports received by the FDA. Enhanced pharmacovigilance with real-world evidence, such as that provided through social media, is essential in protecting public health (Fig. 1). MedWatch, established by the FDA, is designed to expedite and broaden the voluntary reports of serious adverse drug events (ADEs) by healthcare providers and manufacturers.
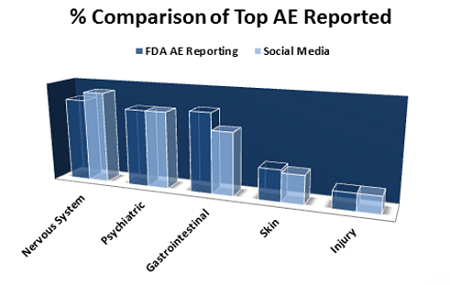
Figure 1: Social networks detecting adverse events (AEs)
Integration of the FDA’s Facility Evaluation and Inspection Program for Human Drugs: A Concept of Operations would apply information across various regulatory departments, the manufacturing site, and organizational intelligence to ensure risk-based facility evaluation decisions. The regulators’ review of submission data and decision making requires the support of adequate data-driven technologies to meet the goals of the 21st Century Cures Act.1 The regulatory guidance developed and adopted by the agency calls for extensive utilization of the continued knowledge as an enabler (ICH Q10).8 Post-marketing surveillance data is maintained by the regulator to monitor the ADE trends. A well-established knowledge management solution is therefore essential to support any well-integrated regulatory body. Technological advancements, including the use of real-time data, predictive modeling, and artificial intelligence, are being applied and are the obvious next steps in meeting the regulators’ goal of promptly delivering safe and effective lifesaving drugs to the patient population. The level of application of such tools varies in each sector; however, the regulators’ acceptance of advancements in modern technologies will accelerate the application of data solutions, artificial intelligence, and other technologies.
Data Analytics
From drug development to market launch, the estimated timeline for a new drug product is 12 years.9 Drug screening itself can take up to five years prior to Phase 1 clinical trials. The probability of a selected drug coming to market is less than one in 10, based on the current trend — not a profitable model. The R&D discovery sector is therefore applying high-tech solutions to enhance productivity and streamline the drug discovery process. Harnessing the advantages of data-driven predictive modeling will enhance the probability of a drug candidate advancing to market, saving valuable testing time and expenses. Such solutions, which have already had an impact on other industries, are now being extensively applied in the discovery segment, including, for example, at Exscientia and Numearate. Major technology providers have collaborated with drug firms such as GSK and Sanofi to deliver high-quality data-driven drug design solutions, dramatically increasing R&D productivity.
Model-based drug development (MBDD) with physiologically based pharmacokinetic (PBPK) or pharmacokinetic pharmacodynamic (PKPD) modeling and simulation solutions is the new trend in drug development. For example, PKPD modeling in early development improves efficiency of dosage delivery and application review; additionally, it allows for predicting bioavailability, PK and PD, while bridging the formulations, populations, and regions. The assessment of variability in response among various subjects helps in managing the risks better. The software tool predicts: drug absorption, metabolism, disposition form structure and physio-chemical properties, PK analysis, PKPD, parameter estimation, design, simulations, etc. These predictions of human PK from animal studies, drug properties, and in vitro-in vivo correlation (IVIVC) are highly beneficial, as models can be built for exposure response from preclinical and animal studies. Further, the human dose can be selected based on the preclinical ER and PK predictions. Real-world data access in clinical trials can influence in various respects: creating the study hypothesis, improving the study design, optimizing data analysis, collaborating and increasing participation in the studies making it most effective and optimal. New tools such as the OpenTrials database10 enable using the available clinical data to develop better conclusions. The ability to capture vast clinical knowledge through new technologies and the regulators’ acceptance of the data sources have a profound impact on the clinical trials space, compelling researchers and regulators to adopt a new mind-set for better patient outcomes.
Process modeling tools are essential to the design and optimization of manufacturing processes. Industry efforts such as that of the System-based Pharmaceutical (SbP) Alliance11 help in developing end-to-end digital design of drug products and their manufacturing processes. Predictive modeling software simulates biopharmaceutical operations in such a way that manufacturing trials can be conducted virtually within the system. This allows for reductions in formulation development costs by eliminating expensive raw materials and production costs. DoEs are executed with multiple controlled variables to identify and determine the desired parameters. During development of the design space, such scientific experiments can be run in multitudes without the application of statistical tools to define the most rational and cost-effective studies required. DoE is the basis of effective QbD development and hence needs to be scientifically justified. Data-driven objective risk assessment has now become part of every formulation and process development stage thus necessitating standardized tools for the same. The advancement of process analytical technology (PAT)12 has allowed for chemo-metric modeling and signal response to be used as a feedback mechanism to optimize process controls, achieving the desired quality attributes. Predictive maintenance (PdM) programs optimizing uptime, costs, productivity, and quality are now being adopted in the biopharmaceutical industry to address the current low overall equipment effectiveness (OEE) in this sector. Vertical integration of production systems enabling intelligent data-driven decision making is becoming the norm. The integration enables efforts such as Industry 4.013 using the information from ERP systems, manufacturing execution systems, supervisory controls, sensing and actuation units, operators, and materials. Technology solutions are essential for collection of process parameter data, quality attributes, and multivariate analysis of that data to detect drifts and incite action. The product/process life cycle data can further be utilized for similar product/process development as well as to eliminate costly developmental studies. The adoption of new data-driven technologies has been identified as beneficial for the industry in minimizing process variability, maintaining a predictable supply chain, and improving product quality, prompting drug firms to adopt such strategies. Application of data-driven automation solutions will help in reducing failures and lead times.
Conclusion
The use of technological advancements, including real-time data, predictive modeling, and artificial intelligence, is becoming a norm in the biopharmaceutical industry. The regulatory acceptance of such data-driven tools has improved the industry’s confidence in using them to develop safe and effective drugs in an optimal development period, and the clear benefits realized by the industry and regulators are helping propel more advancement. Major organizations such as Novartis are now developing strategies to integrate technologies and calling themselves medicines and data science organizations. The prospect of cross sector convergence of intelligence provides opportunities to apply data in a meaningful format for use across sectors, eliminating the costs and time involved in inefficient stand-alone assessments. The second part of this article discusses this convergence, and development of cross sector intelligence (CSI), which enables intelligent decision making based on data sets from multiple sectors.
Note: This article was prepared by the authors in their personal capacity. The opinions expressed are the authors’ own and do not reflect the view of their employer, government, or any agency with which they are affiliated.
References:
1.Public Law 114-255, 21st Century Cures Act, 114th Congress (2015-2016). https://www.gpo.gov/fdsys/pkg/PLAW-114publ255/pdf/PLAW-114publ255.pdf
2.US FDA, Patient Engagement Collaborative. https://www.fda.gov/ForPatients/PatientEngagement/ucm506248.htm
3.Applied Clinical Trials Editors (2012), FDA Clears IND For Clinical Trial Protocol Developed Using Crowdsourcing, Applied Clinical Trials. http://www.appliedclinicaltrialsonline.com/fda-clears-ind-clinical-trial-protocol-developed-using-crowdsourcing
4.U.S. FDA (2016), Best Pharmaceuticals for Children Act and Pediatric Research Equity Act. https://www.fda.gov/downloads/ScienceResearch/SpecialTopics/PediatricTherapeuticsResearch/UCM509815.pdf
5.International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (2009), ICH Q8 (R2), Pharmaceutical Development, Aug. 2009.
6.Alsmeyer, D., Pazhayattil, A. (2014), A Case for Stage 3 Continued Process Verification, Pharma Manufacturing Journal, May 2014. https://www.pharmamanufacturing.com/articles/2014/stage3-continued-process-verification/
7.Kopcha, M. (2017), Continuous Manufacturing: Common Guiding Principles Can Help Ensure Progress, FDA Voice.
8.International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (2009), ICH Q10- Pharmaceutical Quality System, April 2009.
9.Torjesen, I. (2015), Drug development: The Journey of a Medicine from Lab to Shelf, The Pharmaceutical Journal. https://www.pharmaceutical-journal.com/publications/tomorrows-pharmacist/drug-development-the-journey-of-a-medicine-from-lab-to-shelf/20068196.article
10.Grant, B. (2016), Clinical Trial Database Launches, The Scientist. https://www.the-scientist.com/?articles.view/articleNo/47240/title/Clinical-Trial-Database-Launches/
11.GEN (2014), Pfizer, PSE Forge Systems-Based Pharmaceutics Alliance, GEN News. https://www.genengnews.com/gen-news-highlights/pfizer-pse-forge-systems-based-pharmaceutics-alliance/81249676
12.Ciurczak, E. W. (2015), Advances in Process Analytical Technologies, Pharma Manufacturing. https://www.pharmamanufacturing.com/articles/2015/advances-in-process-analytical-technologies/
13.Pharmaceutical Technology Editors (2017), Pharma Industry 4.0 Interface Connects Machines and Automation Systems, Pharmaceutical Technology. http://www.pharmtech.com/pharma-industry-40-interface-connects-machines-and-automation-systems
About The Authors:
 Ajay Babu Pazhayattil has held key management roles with brand name, generic, and contract manufacturing organizations. He is an industrial pharmacist successful in delivering tangible results across multiple segments of pharmaceutical operations. Ajay has extensive experience in conducting global GMP audits and has been effective in conceiving, implementing, and promoting novel technology innovations based on sound scientific principles. His experience extends through solid-dose, liquids, and small and large volume parenteral dosage forms. Ajay is involved with industry organizations and has published multiple journal articles. Connect with him on LinkedIn.
Ajay Babu Pazhayattil has held key management roles with brand name, generic, and contract manufacturing organizations. He is an industrial pharmacist successful in delivering tangible results across multiple segments of pharmaceutical operations. Ajay has extensive experience in conducting global GMP audits and has been effective in conceiving, implementing, and promoting novel technology innovations based on sound scientific principles. His experience extends through solid-dose, liquids, and small and large volume parenteral dosage forms. Ajay is involved with industry organizations and has published multiple journal articles. Connect with him on LinkedIn.
 Marzena Ingram is a pharmaceutical industry professional with extensive quality assurance and technical operations experience; she holds a senior management position. Ingram developed a specialized continued process verification team and spearheaded the implementation of a product life cycle program to meet the global regulatory requirements. She also introduced statistically driven product assessment processes and has been the lead on implementing a comprehensive life cycle management software solution. Ingram also has authored multiple pharmaceutical journal articles. Connect with her on LinkedIn.
Marzena Ingram is a pharmaceutical industry professional with extensive quality assurance and technical operations experience; she holds a senior management position. Ingram developed a specialized continued process verification team and spearheaded the implementation of a product life cycle program to meet the global regulatory requirements. She also introduced statistically driven product assessment processes and has been the lead on implementing a comprehensive life cycle management software solution. Ingram also has authored multiple pharmaceutical journal articles. Connect with her on LinkedIn.
 Naheed Sayeed-Desta has been responsible for providing strategic directions on life cycle management for over 300 active solid-dose products. Naheed champions delivery of science- and risk-based approaches from traditional to novel manufacturing technologies. She is a proven leader in pharmaceutical manufacturing science and technology. Her expertise in providing pragmatic solutions for manufacturing operations is well recognized. She has been the lead author of journal articles. Naheed is an active contributing member of pharmaceutical industry organizations. Connect with her on LinkedIn.
Naheed Sayeed-Desta has been responsible for providing strategic directions on life cycle management for over 300 active solid-dose products. Naheed champions delivery of science- and risk-based approaches from traditional to novel manufacturing technologies. She is a proven leader in pharmaceutical manufacturing science and technology. Her expertise in providing pragmatic solutions for manufacturing operations is well recognized. She has been the lead author of journal articles. Naheed is an active contributing member of pharmaceutical industry organizations. Connect with her on LinkedIn.