
Genentech Masters Breakthrough Therapy Designation
By Ed Miseta, Chief Editor, Clinical Leader
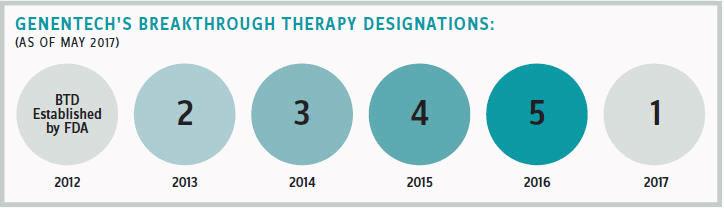
Genentech has garnered 15 Breakthrough Therapy Designations for its medicines since 2013, which is more than any other company. Jeffrey Siegel, senior group medical director for Genentech, believes this success reflects the company’s focus on developing new approaches to address unmet medical needs.
It also makes him a bit of an expert in understanding the intricacies of the FDA program. Siegel was with the FDA for 14 years before joining Genentech and has devoted most of his career to facilitating the development of products to address unmet needs.
“I think the best way to understand the benefits of Breakthrough Therapy Designation is to think about what companies hope for when they approach the FDA with a new product and what they fear might happen over the course of the process,” he says. “What companies generally hope for is that they’ll be able to get concurrence with the FDA on design of the trial, and specifically about the primary endpoint that would determine success or failure of the study. Companies want to know their approach will stand up to the scrutiny of an FDA approval.”
Unfortunately, that process does not always go as planned. When it doesn’t, reaching agreement with the FDA on the design and conduct of the study can be a lengthy and time-consuming process. Just scheduling a meeting with FDA to get feedback can take a long time. Fortunately, this is where Breakthrough Therapy Designation can help.
The designation is a special status the FDA confers on programs believed to address an unmet medical need. This can be a product that has shown benefits in a disease where there are no approved therapies, or it can involve a therapy where data indicates the efficacy is over and above the best available approved therapy.
“For Breakthrough Therapy Designation to apply, clinical data is required to support the efficacy bar set by the FDA,” says Siegel. “In other words, if you have really good data from animal models or other preclinical settings, that’s great. But it won’t get you Breakthrough Therapy Designation. There has to be patient evidence that the treatment provides a particularly high level of benefit.”
DESIGN PROPER ENDPOINTS
When the FDA grants Breakthrough Therapy Designation, it will bring in senior management to assist the sponsor in moving the medicine forward in the quickest manner possible. The FDA agrees to have meetings when the company requires interaction. The designation also means the sponsor gets expedited review once the package is submitted for approval. This could shave several months off the standard approval timeline.
When Siegel was with the FDA, he worked on rheumatoid arthritis at a time when the first biologics were being developed. He helped develop an industry framework for what sponsors would need to show to get approval and demonstrate additional improvement in progression of disease.
“I had a philosophy when I worked at the FDA as a reviewer and clinical team leader,” he says. “Occasionally companies would come forward with programs we felt were not acceptable. When that happened, I instructed the reviewer working under me to always propose an alternative. For example, if the company proposed a laboratory outcome that didn’t necessarily correlate with a clinical benefit, I would work with the reviewer to discuss how the company could show a clinical benefit.”
At Genentech, he continues to work in many of the same areas, still devising endpoints that will be acceptable to the FDA. His advice when designing endpoints? Always look at things from the other person’s point of view. Look at it from the patient’s and FDA’s points of view. Both will be concerned about showing a clinical benefit, not just a benefit on some laboratory endpoint that may provide a benefit to patients.
![]() "If you have really good data from animal models or other preclinical settings, that’s great. But it won’t get you Breakthrough Therapy Designation."
"If you have really good data from animal models or other preclinical settings, that’s great. But it won’t get you Breakthrough Therapy Designation."
 "If you have really good data from animal models or other preclinical settings, that’s great. But it won’t get you Breakthrough Therapy Designation."
"If you have really good data from animal models or other preclinical settings, that’s great. But it won’t get you Breakthrough Therapy Designation."Jeffrey Siegel
Senior Group Medical Director, Genentech
When devising endpoints that show benefits to patients, there are certain things Siegel says are important to consider, especially when there is no established approval pathway. A medicine might meet a biomarker endpoint that doesn’t correlate with clinical benefit. Outcome measures that clearly indicate benefit to patients will generate a positive response from the FDA. When sponsors propose a biomarker as a primary endpoint, they should provide the FDA with persuasive evidence that a change in the biomarker predicts a change in a meaningful clinical outcome.
THE FOCUS MUST BE ON QUALITY
Genentech’s 15 Breakthrough Therapy Designations have come at various points in the development cycle. The earliest time a sponsor can receive the designation is when it first has clinical data from patients showing the product benefits over and above available therapies.
According to Siegel, that can potentially be after Phase 1 in certain areas such as oncology. In immunologic diseases it would more commonly occur after Phase 2. Sometimes it may even come as late as the end of a Phase 3 trial. Regardless of when the submission is made, Siegel believes the single most critical factor to getting the designation from the FDA is data quality. That makes quality data the single greatest challenge as well.
In discussing quality, Siegel likes to use scleroderma as an example. Scleroderma is an immunologic disease where patients develop fibrosis of internal organs as well as thickening of the skin. The fibrosis will lead to damage of the lung, kidney, and other internal organs.
“The most common aspect, and the one that is most apparent, is the skin thickening,” says Siegel. “That can make it difficult for patients to carry out daily living activities and can lead to disability. When we first started our program of tocilizumab (Actemra) for this disease, we wanted to do a Phase 2 proof-of-concept study. For diseases where there is no clear path forward, a typical Phase 2 proof-of-concept study normally takes the form of an open-label study of, say, 25 patients. The goal is to see if responses in the patient indicate a benefit from the drug.”
OPEN LABEL CAN FALL SHORT
With immunologic diseases such as scleroderma, symptoms in a patient will tend to wax and wane. This can create problems for researchers. If you conduct an open-label study in 20 patients and they show improvement, you will not know if there is clinical benefit from the drug, or if that is what might have happened anyway.
“You want to present FDA with quality data, but this situation can leave uncertainty about success of the study,” notes Siegel. “That uncertainty in Phase 2 will also make it more difficult to plan for a Phase 3 study. We were aware of that, so when we began our Phase 2 study we opted to make it a large, randomized trial comparing tocilizumab to a placebo.”
Skin score, which has been validated in scleroderma, was used as the primary endpoint. But Genentech also measured exploratory endpoints for scleroderma that would indicate potential benefit for the serious internal organ manifestations. The results were promising for both easing the skin thickening as well as reducing the rate of lung disease progression. The data was presented to the FDA, and the breakthrough therapy designation was granted. The FDA agreed with Genentech that tocilizumab was a promising new therapy for patients.
FDA regulations state the agency approves drugs based on substantial evidence of efficacy. FDA personnel will tell you that typically means data from one or more adequate and well-controlled trials. A blinded, randomized trial provides that substantial evidence. An open-label trial, in some cases, may not.
“Randomized trials allow researchers to see benefits with the study drug when compared to what patients would have experienced if they’d been treated with standard of care,” says Siegel. “The FDA accepts that as substantial evidence.”
ENSURE SITES ARE TRAINED
In the scleroderma trial, the primary endpoint was what’s called a modified Rodnan skin score. To gather the needed data, an experienced investigator will pinch the skin of a patient in many areas of the body. The researcher determines if the skin has normal suppleness that skin ordinarily has, or if it’s hard and difficult to pinch.
The test is a validated outcome measure and has been shown to correlate with the amount of fibrosis in the skin. Properly trained investigators in studies have been shown to be able to differentiate people who improved, worsened, or experienced no change.
The other endpoint was the measure of lung progression, calculated using the Forced Vital Capacity (FVC). This is a measure of the capacity for gas exchange in the lung. Scleroderma patients with fibrosis of the lung will have decreasing FVC measures.
While the FVC measure is fairly straightforward, recruiting investigators who aren’t adequately trained on the Rodnan skin score can create problems. The test needs to be conducted in a uniform manner so as to generate successful readouts at the end of the study.
“To get high-quality data, we do everything possible to create uniformity among the investigators,” states Siegel. “In the case of scleroderma, this is particularly important. A researcher in Paris and another in LA might perform the same pinch test for skin fibrosis, but interpret the results differently.”
To avoid this possible tainting of data, Genentech brought together the targeted investigators to make sure they were adequately trained on how to properly read patients and to ensure everyone was performing the test the same way.
UNIFORMITY ACROSS STUDY RESULTS
Genentech retained the services of a couple of academic investigators who were expert clinicians and had worked together to standardize the Rodnan skin score. They were able to train investigators on the exact way to apply the Rodnan skin score and make sure everyone was interpreting the results accurately.
“It was a great way for us to get uniformity across our study results,” says Siegel. “This is something everyone should be aware of when conducting proof-of- concept studies, particularly Phase 3 studies where you tend to have clinicians in multiple countries. Conducting the training is a challenge, but the rewards you reap are huge.”
In conducting the training, a video is often sufficient. That is generally the case if investigators are familiar with the test and have conducted it in the past. The video serves as a refresher on what they need to do. For this study, the expert clinicians were able to offer additional insights to investigators. To ensure everyone had an opportunity to interpret the results, the clinicians also brought in patients who were willing to participate in the training.
“The patients would meet with individual clinicians who would then perform the same tests,” adds Siegel. “They were asked to score the hands, arms, chests, and legs. The experts then checked to see if all clinicians scored each of those body parts the same way.”
Some discrepancies were noted. When that happened, the clinicians were brought together to talk about how they were scoring patients. A consensus was reached on how to apply standardized criteria to see that all patients were scored the same way.
“By bringing good science, quality data, and a focus on unmet medical need to the FDA, companies can make major advancements in unmet medical needs,” adds Siegel. “Attaining a Breakthrough Therapy Designation might require some additional insight and work, but by working with the FDA in this manner, we will be able to bring our therapies to patients more quickly and efficiently.”