
A Road Map To China's Medical Device Registration Process
By May Ng and Ren Dazhi, ARQon

China has one of the most promising medical markets and its healthcare industry is currently the second largest in the world, following closely behind the U.S. From 2015 to 2019, the CAGR for the Chinese medical devices market has increased significantly, and it is expected to continue rising even more in the next four years due to the growing population, the escalating prevalence of diseases and disorders, and many other factors. Therefore, the Chinese medical device market is highly sought after.
Medical device approval is required to be obtained from the National Medical Products Administration (NMPA) (see ARQon’s website here for more information). The pre-market or product registration procedures are different for imported products and local domestic products.
Registration For Imported And Domestic Medical Devices
For overseas manufacturers who are importing their medical devices to China, you will need to obtain a product registration license. For domestic products, both the product registration license and production approval license are mandatory.
A total of three levels are responsible for the management of Chinese medical devices, namely the NMPA, the provincial bureau, and the municipal bureau. Each of their respective responsibilities/jurisdictions can be seen in the table below.
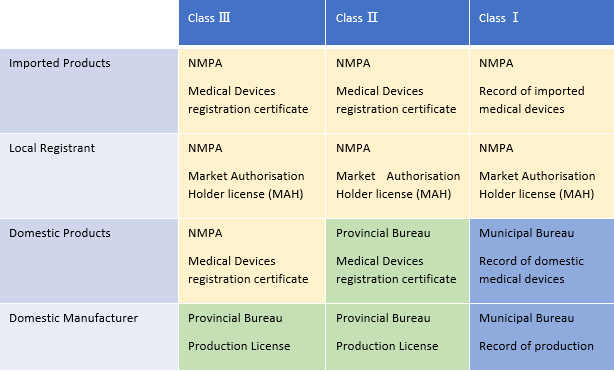
Table 1: Responsibilities of the central and local authorities. Courtesy of ARQon.
NMPA's responsibility is:
- To review and approve registration of class 2 and class 3 imported medical devices.
- To receive the record of class 1 imported medical devices.
- To organize the inspection of foreign manufacturers' QMS.
- To review and approve domestic class 3 medical devices.
The provincial bureau, such as Zhejiang Medical Products Administration, is responsible for:
- To review and approve class 2 domestic medical devices.
- To review and approve domestic manufacturers' production license for medical devices of class 2 and class 3.
The municipal bureau’s responsibility is:
- To receive the record of domestic class 1 medical devices.
- To receive the record of production of domestic manufacturers, which only produce medical devices of class 1.
For class 2 and class 3 medical devices, foreign manufacturers will obtain one license, which is the medical devices registration certificate. Domestic manufacturers will obtain two licenses, which are the medical devices registration and the production license.
For medical devices of class 1, foreign manufacturers are responsible for one kind of record, and domestic manufacturers are responsible for two kinds of records.
Foreign manufacturers are required to have a local representative company to be the marketing authorisation holder (MAH) for the submission of their medical device registration.
There may be some special situations in which the MAH requirement is applicable to domestic manufacturers.
To register an imported medical device, the following steps need to be taken:
- Determine which class the device falls under, and then plan the strategy and timeline for the product registration.
- Start preparing the product technical requirements and reference to various standards: national, industrial, and international. The main content of the technical requirement should include performance, safety, and inspection methods of the finished product.
- When the prototype is ready, move on to the type inspection. The type inspection process will take at least six months to complete.
- Obtain the inspection report and conduct clinical trials. Clinical trials usually take more than a year to complete; however, they are not mandatory for all devices. NMPA has issued a catalogue of medical devices exempt from clinical trials. If the declared product is in the catalogue, provide a comparison report between the declared product and marketed products in China. Note that the declared product should have the same name as the one in the catalogue. During clinical evaluation, the declared product’s similarities and differences should be compared with one or more medical devices of the same variety to prove that they are substantively equivalent. For medical devices that require the conduct of clinical trials, these should be carried out in accordance with the good clinical practice (GCP) guideline. For imported medical devices that are clinically tested abroad, submit the clinical trial data overseas if the clinical trials meet the relevant Chinese regulations and technical requirements.
- Concurrently, you can start the preparation of regulatory documents. Before submitting the application for registration, it is important to compile the registration application materials such as the risk management report.
- For inspection of medical devices that involve performance, medical electrical safety, EMC, and software, companies that are qualified for IEC60601-1, IEC61010, IEC60601-1-2, and IEC62304 overseas are not exempted from the submission of inspection reports that meet the standards of GB9706, GB4793, YY0505, and YY0664 in China due to the differences in versions or contents.
- The bio-compatibility test reports can be accepted if you have previously conducted the ISO10993 experiment overseas, and the laboratory can meet the good laboratory practices (GLP) requirements and the test reports can meet the requirements.
- In the product registration submission, once the application is accepted, the Center for Medical Device Evaluation (CMDE) will need about 60 to 90 workdays to finish the first-time review. Once done, you will receive a corrective notice and you will have to submit the correct patent materials within a year. Exceeding this time frame will lead to a rejection of the application. This emphasizes the importance of preparation for registration. An extra second review will be needed for class 2 and 3 products. The final step before the issuance of a certificate is to get administrative approval, which is usually done within 20 workdays.
Figure 1: Flow Chart for Imported Medical Device Registration
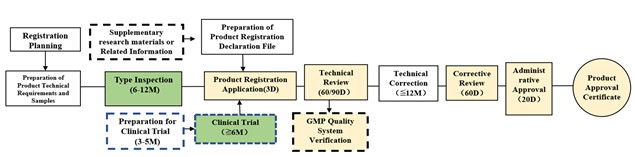
Flowchart courtesy of ARQon.
Remarks:
- “D” means days, “M” means months.
- Boxes shaded in yellow are under NMPA. The technical review period for the registration of imported class 2 medical devices is 60 working days. The technical review period for the registration of imported class 3 medical devices is 90 working days.
- Boxes shaded in green require collaboration with a testing institute or a clinical institution.
- Boxes shaded in white need to be completed by the companies themselves.
- Boxes with dotted lines represent events that may not happen.
Figure 2: Flow Chart for Domestic Medical Device Registration
Product Registration
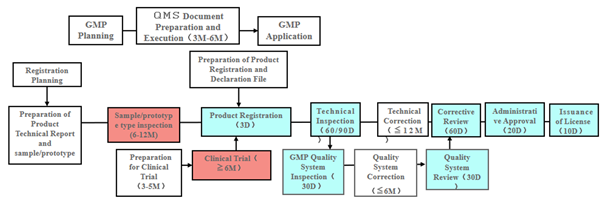
Flowchart courtesy of ARQon.
Remarks:
- “D” means days, “M” means months.
- Boxes shaded in blue are under NMPA. The technical review period for the registration of imported class 2 medical devices is 60 working days. The technical review period for the registration of imported class 3 medical devices is 90 working days.
- Boxes shaded in red require collaboration between a verification institute and a clinical institution.
- Boxes shaded in white need to be completed by the companies themselves.
Figure 3: Domestic Manufacturer License
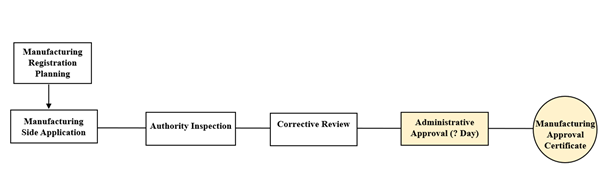
Remarks: 20 working days for the manufacturing license approval
The authority fees for the respective steps can be seen in the table below.
|
Steps: |
Prices (€): |
|
Initial registration for class 3 products |
39,000 |
|
Initial registration for class 2 products |
26.500 |
|
Registration alterations for class 3 products |
6,300 |
|
Registration alterations for class 2 products |
5,300 |
|
Registration renewals for class 3 and class 2 products |
5,000 |
|
Application for clinical trial approval (only for high-risk medical devices) |
5,400 |
Table 2: Registration fees
Fast-track Special Approval Channel For Medical Devices
There are two types of special approval channels available:
- Innovative medical device review process
- Medical device priority approval process
Innovative Medical Device Review Process
The innovative medical device review process was implemented in hopes that it can accelerate the regulatory approval process for innovative medical technology. The whole process takes about 73 workdays for imported products and an additional 20 workdays for domestic products due to the need for provincial administration.
Medical devices that meet the following criteria per NMPA are classified as innovative medical devices:
- Invention patents
- The applicant legally owns the patent right to the product’s core technology invention in China
- The applicant legally obtains the invention patent right or its right to use it in China through transfer and the application date for the special review of the innovative medical device is no more than five years from the patent authorization notice.
- The application for the core technology invention patent has been published by the patent administration department under the state council, and a search report is issued by the patent search and consultation center of the state intellectual property office. The report states that the core technology of the product is innovative and creative.
- The applicant has completed the preliminary research of the product and has the basic prototype. The research process is real and controlled, and the research data is complete and traceable.
- The main working principle or mechanism of the product is the first case in China. The product performance or safety is fundamentally improved compared with similar products. The technology is at the international leading position and has significant clinical application value.
Among the various benefits you can gain from this review process, the main benefit is the ability to establish good early communication with the review agency. Before the product registration application is accepted and during the technical review process, CMDE designates a reviewer to communicate and provide guidance in a timely manner at the request of the applicant to discuss relevant technical issues.
Medical Device Priority Approval Process
The medical device priority review and approval process was implemented on Jan. 1, 2017. Currently, all class 3 medical products are qualified to request consideration under this scheme, but for class 2 devices it is limited to only imported products.
Medical devices that meet the following criteria are applicable to this channel:
- Diagnosing or treating rare diseases with obvious clinical advantages.
- Diagnosing or treating malignant tumors with obvious clinical advantages.
- Diagnosing or treating endemic and common diseases in the elderly for which there are currently no effective diagnosis or treatment.
- Dedicated to children and has obvious clinical advantages.
- Clinically urgently needed and there are no such medical devices approved for registration in China.
A benefit of entering this channel is active communication in accordance with relevant regulations during the technical review process. Furthermore, NMPA gives priority to its administrative approval once the technical review is done.
The table below compares the innovative medical device review and the medical device priority approval procedures.
|
|
Innovative medical device review process |
Medical device priority approval process |
|
Condition |
- Owns innovation patent for the core technology - first product in China - Has significant clinical application value |
- Urgently needed in clinical settings - State key scientific and technological projects |
|
Advantages |
- Consultancy is provided even before registration - Specific person assigned - Priority in review and approval |
- Special communication - Priority in review and approval |
|
Time |
- Before the application for registration |
- Together with the application for registration |
Table 3: Comparison table
Conclusion
As China is one of the major medical device markets in the world, manufacturers may want to consider expanding their products there. It takes a much longer time for foreign manufacturers to obtain medical device approval in China, but companies can consider the fast-track special approval channels for medical devices if the product meets the innovative or priority medical device requirements. Though there is an exemption from local clinical trials for certain products, foreign manufacturers should be mindful that they may still be subject to local clinical trials if new innovations are incorporated into the medical device or if there are safety concerns about the device. A comprehensive regulatory strategy and good communication skills with the Chinese regulators are important, as they can dictate the application requirements and approval timeline.
About the Authors:
 May Ng is the global director of ARQon, Asia Regulatory & Quality Consultancy for medical devices and drugs. She assists companies in product development and product registration in Asia, global approval, and serves as a CE Representative. Previously, Ms. Ng was in the Singapore Health Sciences Authority (HSA) to complete the implementation of the Singapore regulatory framework and approve medical device/IVD and GDP/ISO13485-compliance dealer licenses. Currently, she holds roles as the Medtech Regulatory Advisor for organizations such as NHIC, IPI, A*STAR A*ccelerate ETPL/DxDHub, MOL COVID 19 panel, and NUS Enterprise. She is a committee member of Singapore Manufacturing Federation’s MedTech Industry Group, China-ASEAN, and US-ASEAN medical device planning. She is also the co-chair for ASEANMed. Previously, she was WHO trainer for regulators, assessor for the Prime Minister’s Office for Startup innovation, and regulator representative for the formation of AHWP and ACCSQ MDPWG/ASEAN Medical Device Directive.
May Ng is the global director of ARQon, Asia Regulatory & Quality Consultancy for medical devices and drugs. She assists companies in product development and product registration in Asia, global approval, and serves as a CE Representative. Previously, Ms. Ng was in the Singapore Health Sciences Authority (HSA) to complete the implementation of the Singapore regulatory framework and approve medical device/IVD and GDP/ISO13485-compliance dealer licenses. Currently, she holds roles as the Medtech Regulatory Advisor for organizations such as NHIC, IPI, A*STAR A*ccelerate ETPL/DxDHub, MOL COVID 19 panel, and NUS Enterprise. She is a committee member of Singapore Manufacturing Federation’s MedTech Industry Group, China-ASEAN, and US-ASEAN medical device planning. She is also the co-chair for ASEANMed. Previously, she was WHO trainer for regulators, assessor for the Prime Minister’s Office for Startup innovation, and regulator representative for the formation of AHWP and ACCSQ MDPWG/ASEAN Medical Device Directive.
 Ren Dazhi is on ARQon China’s team to help global companies to obtain product approval of medical device in China. He was a member of the First Executive Committee of the Medical Device Classification Committee, China Food and Drug Administration, First Council of the Medical Device Professional Committee, China Drug Administration Research Association, and Technical Committee of National Medical Clinical Laboratory and Standardization of In-Vitro Diagnostics. Since 2001, he has been engaged in the supervision and management of medical device classification and standard management, testing, GMP quality system, review, and approval. He was responsible for pre-market review and approval of medical devices and post-marketing supervision. He organized the formation of Beijing medical device registration and regulatory documents.
Ren Dazhi is on ARQon China’s team to help global companies to obtain product approval of medical device in China. He was a member of the First Executive Committee of the Medical Device Classification Committee, China Food and Drug Administration, First Council of the Medical Device Professional Committee, China Drug Administration Research Association, and Technical Committee of National Medical Clinical Laboratory and Standardization of In-Vitro Diagnostics. Since 2001, he has been engaged in the supervision and management of medical device classification and standard management, testing, GMP quality system, review, and approval. He was responsible for pre-market review and approval of medical devices and post-marketing supervision. He organized the formation of Beijing medical device registration and regulatory documents.