
Companion Diagnostics Leakage In Oncology — What Biopharma Companies Need To Know

By Alex Vadas and Brian Baranick, L.E.K. Consulting
Biopharmaceutical companies continue to adopt personalized medicine approaches that leverage patient-specific biomarkers to stratify patients. Companion diagnostic (CDx) tests are indispensable to personalized medicine, which relies on accurate, reliable, and clinically meaningful tests to identify the appropriate drugs for each patient.
The commercialization of these tests raises challenges for biopharmaceutical companies, which strive to provide patients access to drugs. Biopharmaceutical companies typically partner with global in vitro diagnostic (IVD) companies to complete the steps that lead to a successful CDx test: development, regulatory approval (often sought during the drug’s pivotal trial), commercialization, and widespread distribution into laboratories. Local or central laboratories often process collected samples, perform the tests (from the IVD partner), and report test results back to physicians. Laboratories might also create their own “homebrews,” or laboratory-developed tests (LDTs), to reduce input costs.
Most biopharmaceutical companies that launch products with companion diagnostics inevitably face CDx-related patient leakage, or the failure of eligible biomarker-positive patients to receive appropriate targeted therapy. Patient leakage occurs throughout the test workflow from the collection of a sample/biopsy through the receipt of an actionable test report (see Figure 1). While sample access issues are well known (especially in sample constrained environments such as NSCLC), this article examines issues during test ordering, test performance, and results reporting that can lead to patient leakage.
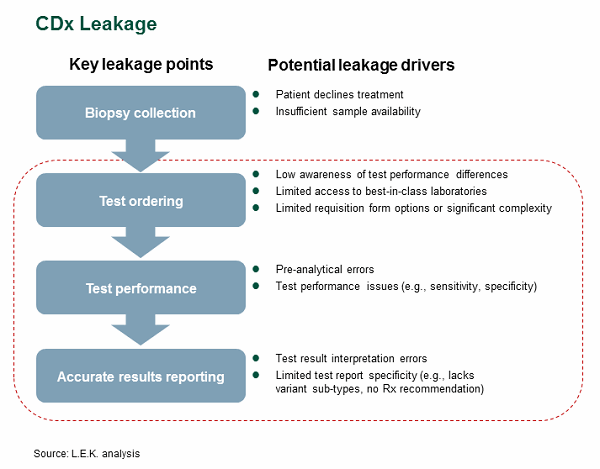
Figure 1
Test Ordering Issues
Most physicians — especially outside of oncology — have a limited understanding of the technical nature of laboratory medicine. They often rely on their local/national labs to offer the appropriate testing and testing algorithms, select the best technologies, and report results accurately. Although labs have patients’ best interests in mind, it may not be feasible for them to offer the most ideal test or testing algorithm.
A patient’s failure to receive the appropriate test or most comprehensive testing algorithm for a given biomarker is a key source of patient leakage. We have found that two converse factors drive ordering-related patient leakage: (1) the availability of too many options, which makes it difficult for physicians to choose the most appropriate option for a given patient situation, and (2) the availability of too few options, which may not include the optimal one. One of the leading national reference labs is an example of a lab that offers a vast array of options — in this case, multiple tests and algorithms for HER2, which is used as a companion diagnostic for Herceptin (see Figure 2).
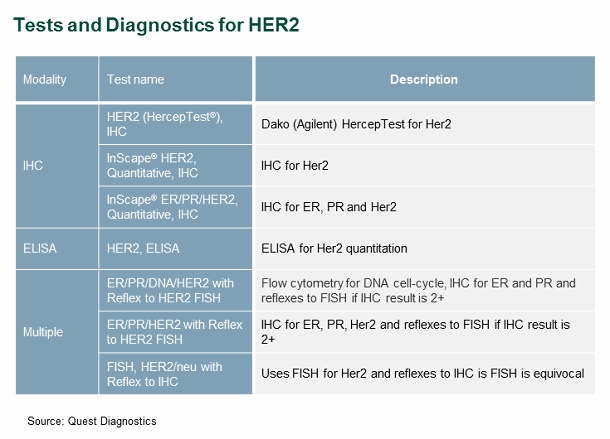
Figure 2
This vast array of testing options is ideal only if the ordering physician has in-depth knowledge of laboratory medicine, and as previously stated, most do not. Inappropriate test ordering may lead to sub-optimal HER2 tests or algorithms, and thus subsequent false negatives, thereby preventing eligible patients from receiving beneficial therapy.
Test Performance Issues
Assuming the optimal test and algorithm were selected, patient leakage can still occur due to variance in analytical test performance. Analytical test performance often varies by test modality (e.g., PCR, IHC, FISH) and by vendor, and a broad array of technical factors (e.g., variant coverage, probe specificity, antibody sensitivity) can influence such performance. Peer-reviewed studies suggest that sensitivity-related issues can lead to false negative rates of 4 to 10 percent for approved molecular diagnostic tests, but our research has observed situations in which a confluence of factors can make the false negative as high as 25 percent.
Pathology tests (e.g., IHC, FISH) may also face additional interpretation challenges, given that these tests’ results are often subjective. Test interpretation typically requires a highly skilled and trained pathologist, but pathology tests’ subjective nature can lead to significant variation between pathologists and labs. In an effort to reduce interpretation variance, most IVD manufacturers often develop “test interpretation manuals,” which provide a form of test interpretation standardization (see Figure 3).
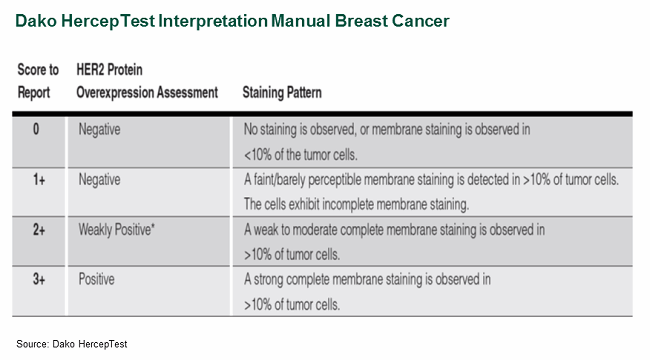
Figure 3
Some studies have attempted to quantify the true impact of subjective interpretation, reporting false-positive rates as high as 26 percent for locally tested samples retested by central labs, where pathologists may be more familiar with the tests. A recent study found that 4 percent of locally tested negative HER2 patients were actually found to be positive upon central lab testing, suggesting that approximately 7,000 U.S. patients were not receiving appropriate HER2 therapy.1
Results Reporting Issues
Test reporting forms may contribute to patient leakage and inadvertently lead a physician to misunderstand a patient’s true biomarker status or make sub-optimal therapy selection decisions. Reporting can be heterogeneous, with some reports including a simple “biomarker negative/positive” (i.e., EGFR mutation positive or EGFR mutation negative), or it can provide mutation-specific differences (e.g., EGFR positive with exon 19 deletions) within a given biomarker.
As a case study, we investigated EGFR in lung adenocarcinomas. In this specific context, a simple EGFR negative/positive report is insufficient to make informed therapy selection decisions. Studies have shown that susceptibility to the tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib can vary greatly depending on the specific EGFR mutation pattern (see Figure 4).
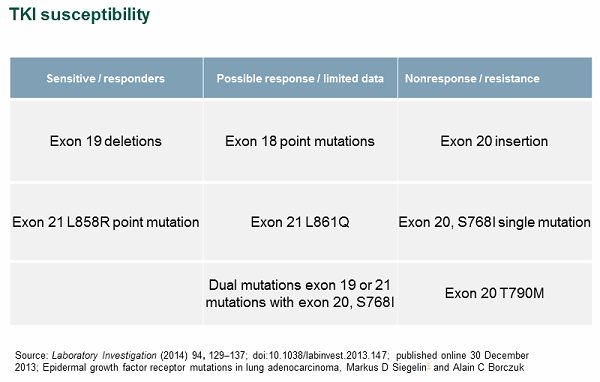
Figure 4
In the case of EGFR, it is important that labs use a testing modality capable of interrogating the various mutations. It is equally important for the associated report to provide, at a minimum, the mutation status for each specific EGFR mutation. Ideally, the report should arm the physician with the medical information needed to make informed therapy selection decisions.
We have not seen specific studies that quantify the impact of high-quality lab reports on patient leakage, but our experience suggests that a meaningful percentage of patients can be lost due to variance in lab reporting.
It Will Get Better and Worse Going Forward
The industry is working to improve the situation in the following ways:
- Increasing genetic education in medical schools and through continuing medical education
- Adopting technologies with very high sensitivity (e.g., NGS, digital PCR), which can improve sensitivity
- Using technologies that have broad variant coverage (e.g., NGS)
- Adopting digital pathology solutions that help standardize interpretation
- Increasing regulatory oversight and guidance
Despite these efforts, we expect continuing increases in number of biomarkers, number of test offerings, number of test algorithms, and overall test complexity to lead to more patient leakage during test ordering, test performance, and results reporting.
Potential Solutions
Biopharma companies need to be aware that launching a drug with a regulatory-approved CDx test does not guarantee access. Even with a successful commercialization strategy, a CDx test is not necessarily immune to patient leakage due to test ordering, test performance, and results reporting issues, which can lead to lower-than-expected drug revenues and reduced clinical benefit for patients. Potential strategies to minimize patient leakage and maximize access to drugs with a CDx may include:
- Characterizing and quantifying leakage points and how they vary by situation (care setting, region, patient-specific)
- Identifying contributing forces to leakage and developing plans to address those forces by stakeholder (e.g., pathologist, physician, payer, provider, patient)
- Developing commercial strategies to address leakage points and contributing factors
- Developing CDx lifecycle strategies
- Outlining new partnering strategies that can help reduce patient leakage
References:
- Kaufman et al., Cancer 2014; 120:2657-64
About The Authors:
 Alexander Vadas, Ph.D., is a managing director and partner in L.E.K. Consulting’s Biopharmaceuticals & Life Sciences practice. He joined L.E.K. in 2000 and focuses on diagnostics, research tools, and personalized medicine. Within those areas, Vadas has worked with a range of established and emerging clients in the areas of corporate strategy, product strategy, and planning and transaction support. He received both his B.S. and Ph.D. degrees in chemical engineering from the University of California, Los Angeles.
Alexander Vadas, Ph.D., is a managing director and partner in L.E.K. Consulting’s Biopharmaceuticals & Life Sciences practice. He joined L.E.K. in 2000 and focuses on diagnostics, research tools, and personalized medicine. Within those areas, Vadas has worked with a range of established and emerging clients in the areas of corporate strategy, product strategy, and planning and transaction support. He received both his B.S. and Ph.D. degrees in chemical engineering from the University of California, Los Angeles.
 Brian Baranick is a managing director and partner in L.E.K. Consulting's Los Angeles office. He joined L.E.K.’s Biopharma and Life Sciences practice in 2007 and has supported clients across many sectors including biopharmaceuticals, life science tools, diagnostics, and personalized medicine. Baranick also has experience across a broad range of therapeutic area and technology segments. He has advised both established and emerging clients in corporate strategy, product strategy, and planning and transaction support. His thought leadership can be found in several L.E.K. Executive Insights, including Personalized Oncology: A Roadmap and Forging a Path From Companion Diagnostics to Holistic Decision Support. Baranick received his Ph.D. in molecular biology from the University of California, Los Angeles.
Brian Baranick is a managing director and partner in L.E.K. Consulting's Los Angeles office. He joined L.E.K.’s Biopharma and Life Sciences practice in 2007 and has supported clients across many sectors including biopharmaceuticals, life science tools, diagnostics, and personalized medicine. Baranick also has experience across a broad range of therapeutic area and technology segments. He has advised both established and emerging clients in corporate strategy, product strategy, and planning and transaction support. His thought leadership can be found in several L.E.K. Executive Insights, including Personalized Oncology: A Roadmap and Forging a Path From Companion Diagnostics to Holistic Decision Support. Baranick received his Ph.D. in molecular biology from the University of California, Los Angeles.