
Decoding FDA's Refuse To Receive (RTR) Standards For ANDA Submissions
By NandaKumar Gollapalli, assistant general manager, Freyr

Generic drugs are a crucial part of the U.S. healthcare system, making up 90 percent of all drug prescriptions dispensed in the country. These medicines have saved patients a tremendous amount of money and have paved a path toward more affordable healthcare.
As part of its agenda to speed accessibility of high-quality generic drugs, the FDA tentatively approved 190 abbreviated new drug applications (ANDAs) in the 2018 fiscal year (FY2018). Despite this progress, the approval rate for ANDA submissions is much lower than expected. In FY2018 alone, the FDA received a total of 1,044 ANDA applications, out of which only 10 percent received first cycle approval.
Though the FDA has laid down clear steps for ANDA submissions, applicants still struggle with the procedural challenges in preparing and submitting their applications. As such, it is important to dig deeper into the process and understand the possible reasons why the FDA may refuse to approve — or even refuse to receive (RTR) — an ANDA submission.
It certainly is important to be first to market when it comes to launching generic drugs, and sponsors and manufacturers are aware of how critical first-mover advantage is in the success or failure of a product. To improve the likelihood that an ANDA will get through the FDA’s review cycle on the very first attempt, it is important for sponsors to submit a complete and scientifically accurate ANDA — issuance of an RTR determination will only add financial burden, delay market-entry, and increase market competition. This article will explain the reasons why the FDA issues RTRs and how sponsors can best prepare an ANDA for success.
An Introduction To Refuse To Receive (RTR)
Put simply, RTRs, which apply both to ANDAs and to prior approval supplements (PASs) to ANDAs (required when making a major manufacturing or other change to a previously approved ANDA), indicate that FDA does not consider the submitted information to be substantially complete. When an ANDA is submitted to the FDA, the agency evaluates each application to ensure that it is complete and contains all the required information as per the section 505 (j) (2) (A) of the Federal Food, Drug and Cosmetic Act (FD&C Act) and doesn’t contain any deficiency as described in 21 CFR 314.101 (a) and (e).
An FDA reviewer can determine that the application is not complete and ready for review, refusing to receive it, based on:
- Inadequate stability data
- Incomplete response to screening deficiency
- Inadequate dissolution data
- Qualitatively (Q1) and quantitatively (Q2) dissimilarity from the innovator drug
- Response to screening deficiency delayed beyond the prescribed time limit.
There are two milestones in the ANDA submission when the agency decides the necessary action to be taken by the applicant with respect to RTR):
- The submission of an application to FDA
- FDA accepting the submission and initiating the application review
Both stages are governed by their respective rules (like stability duration, justification for impurities, missing application form for initial submission, and not responding within 7 calendar days/not fulfilling the FDA requirements during screening deficiency), and applicants must clearly understand the acceptance criteria. If it is ignored, it may lead to application rejection.
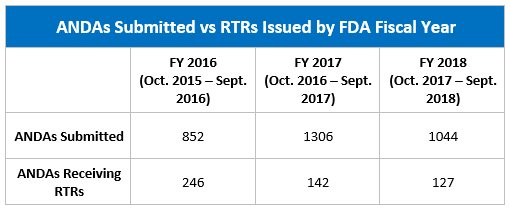
The table below shows the number of ANDAs submitted and the number of those the FDA refused to review from FY 2016 through FY 2018.
RTR Scenarios: Major And Minor Deficiencies
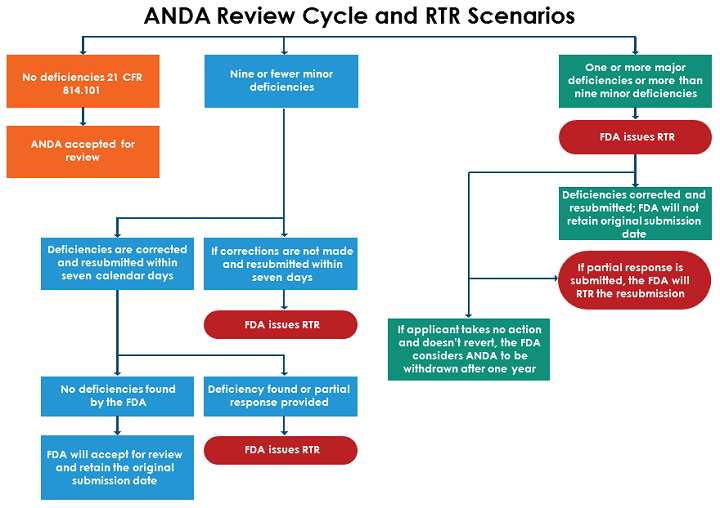
When an ANDA is submitted for review, the FDA will determine if the application is complete from a high-level view. The FDA will check for any missing details, highlight deficiencies (both major and minor), and mark if any corrections are necessary. To ensure an application is correctly completed and filed, it is important to understand the differences between major and minor deficiencies.
- A major deficiency is one the FDA considers “significant in nature, such as some found in 21 CFR 314.101(d) and 314.101(e). If a major deficiency is identified, FDA will RTR the ANDA.
- A minor deficiency is one that the agency considers less critical in nature and can be remedied easily. If the ANDA contains fewer than 10 minor deficiencies, the FDA will notify the applicant via phone, fax, or email and give them seven calendar days to correct these deficiencies or amend the ANDA. If the deficiencies aren’t corrected within seven days or additional deficiencies are found, the agency will issue an RTR.
The figure below shows the FDA’s process for reviewing ANDAs and issuing RTRs, depending on the number and type of deficiencies found.
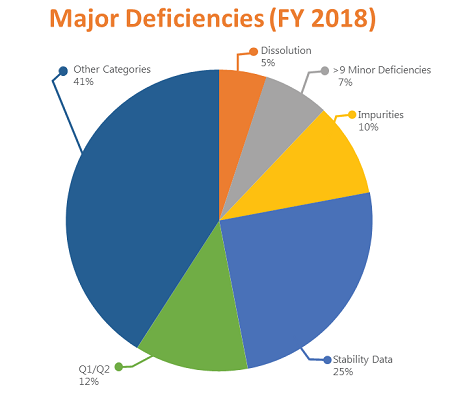
This pie chart shows the distribution of major deficiencies by type in FY 2018:
Deficiencies In Abbreviated New Drug Applications
This section provides example deficiencies, organized by module of the Common Technical Document (CTD), that could lead to an RTR.
Module 1
Minor Deficiencies:
- Incomplete Form FDA 356h, such as
- Field 11: Full chemical name not provided
- Field 20: Patent certification is inconsistent with the patent certification provided in Module 1.3.5.2
- Field 28: Establishment information does not match with the facilities information provided in Modules 3.2.S and 3.2.P
- Field 29: Typo in drug master file (DMF) number or failure to list all the DMFs referenced in module
- Basis for submission 21 CFR §314.94(a)(3)
- Failing to provide the appropriate basis of submission — designated reference listed drug (RLD) and reference standard (RS; if applicable) currently listed in the Orange Book
- If an ANDA suitability petition is required, failure to provide the docket number or FDA’s correspondence approving the petition
- Labeling (Module 1.14)
- eCTD: Legibility of draft and RLD container labels
- Failing to provide the proposed container and carton labels for each strength and each packaging configuration (container size)
- Failing to provide the RLD container and carton label for each strength
Major Deficiencies:
- Unsigned Form FDA 356h
- Failure to submit Form FDA 356h
Module 2
Minor Deficiencies:
- Provide separate PDF and Word documents
- Missing summary data tables in module 2.7
- Failure to provide the certificate of analysis for each strength of the RLD
- Failure to provide the exact location of the long-term storage stability (LTSS) study reports and data (Table 10), along with working hyperlinks to respective information
Major Deficiencies:
- Inadequate dissolution studies, lacking:
- Minimum of 12 units
- Use of FDA-recommended test media
- ½ tablet dissolution for modified-release products with functional score marks
- General deficiencies of in-vitro dissolution (Table 5)
- Not conducted on 12 units
- Not conducted on all strengths (test vs. RLD)
- Not conducted in all test media
Module 3
Minor Deficiencies:
- Lack of legibility in documents/data
- Failure to translate non-English content into the English language
- Failure to follow the ANDA checklist
- Missing batch reconciliation and label reconciliation information
- Executed batch reconciliation tables don’t include theoretical, actual, and packaged yield
- Yield is not expressed in dosage or product units (e.g., number of tablets and bottles, number of vials, etc.)
- Potential impurities not listed in tabular format as per FDA recommendation
Major Deficiencies:
- Failure to demonstrate Q1/Q2 sameness for sterile drug products, ophthalmic, and otic solutions to the RLD
- Lack of justification for unknown/unspecified impurities
- Not providing method validation/verification reports
- Inconsistent functional scoring configuration with RLD
- Lack of compliance with inactive ingredient database (IID) limits for excipients for solid orals/parenterals/opthalmics/otics/topical drug products
- Lack of justification (supporting data and information) for impurities (specified identified or specified unidentified) where proposed acceptance criteria (AC) percentage exceeds qualification threshold (QT) or identification threshold (IT), respectively
- Proposed AC percentage exceeds QT or IT percentage, as applicable
- Failure to provide stability data on two discrete API lots for each strength of drug product, and on a minimum of three drug product batches of each strength
- Failure to provide six months (180 days) of stability data with a minimum three time points
- Accelerated and long-term stability studies
- Intermediate studies for all three batches of the specific strength if accelerated stability study shows significant change or failure of any attribute
- Failure to submit worst-case scenario and non-worst-case stability data related to container orientation
- Lack of verification for all stability start and pull dates
Consequences of an RTR
When the FDA issues an RTR, it is communicating to the sponsor that their ANDA has effectively not been “received,” and it returns 75 percent of ANDA fee already paid by applicant. As the result of an RTR, sponsors face the following ramifications:
- Loss of 25 percent of the ANDA fee
- Loss of original submission date
- Loss of market exclusivity period (180 days) in the case of NCE-1 submissions
- Delay in product launch
If the applicant submits required information and material to correct the deficiencies, the FDA is all open to consider (if found substantially complete) the new and corrected version of ANDA. Upon such new submission, however, the applicant needs to pay the total ANDA submission fee, accordingly. The date of the revised submission will be considered as the new application date.
The applicant can request the FDA for a reconsideration, if the applicant disagrees with the major deficiencies identified and notified by the agency. Then the ANDA applicant can provide the relevant supporting information and material to the FDA and request for reconsideration. If the FDA does not agree even after submission of the supporting information, the applicant can request for a teleconference with the Agency for further evaluations. If the reconsideration issue remains unsolved, the applicant should refer to 21 CFR 314.103 and guidance for industry Formal Dispute Resolution.
Conclusion
There have been several guidance documents issued by the FDA elaborating on the requirements for ANDA filing suitability. In addition, the FDA has published Q&A documents answering questions they have received from industry on these topics. In this current environment of information accessibility, an RTR for an ANDA can be easily avoided. However, it is important for product development, quality and Regulatory professionals to understand every aspect of the FDA’s guidance related to their product and generate necessary data for a high quality ANDA submission for successful and quick review and approvals.
References:
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/anda-submissions-content-and-format-abbreviated-new-drug-applications
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/anda-submissions-refuse-receive-standards-rev2
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/anda-submissions-refuse-receive-lack-justification-impurity-limits
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/andas-stability-testing-drug-substances-and-products-questions-and-answers
- https://www.fda.gov/regulatory-information/search-fda-guidance-documents/controlled-correspondence-related-generic-drug-development
- https://www.fda.gov/media/107325/download
- https://www.fda.gov/media/107637/download
 About The Author:
About The Author:
NandaKumar Gollapalli is a regulatory professional with 15+ years’ experience in handling end-to-end regulatory services for regulatory strategy, registration, and life cycle management for innovator and generic products to US FDA.