
FDA Updates Several 510(k) Guidance Documents
By Mark Durivage, Quality Systems Compliance LLC
The U.S. Food and Drug Administration (FDA) Center for Devices and Radiological Health (CDRH) and Center for Biologics Evaluation and Research (CBER) recently issued four final guidance documents related to 510(k) regulatory submissions:
Biologics Evaluation and Research (CBER) recently issued four final guidance documents related to 510(k) regulatory submissions:
- The Special 510(k) Program - Guidance for Industry and Food and Drug Administration Staff
- The Abbreviated 510(k) Program - Guidance for Industry and Food and Drug Administration Staff
- Format for Traditional and Abbreviated 510(k)s - Guidance for Industry and FDA Staff
- Refuse to Accept Policy for 510(k)s - Guidance for Industry and Food and Drug Administration Staff
The Special 510(k) Program
The Special 510(k) Program, published Sept. 13, 2019, supersedes the Special 510(k) content in The New 510(k) Paradigm - Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications, issued March 20, 1998. This program provides device manufacturers an “optional pathway for certain well-defined device modifications where a manufacturer modifies its own legally marketed device, and design control procedures produce reliable results that can form, in addition to other 510(k) content requirements, the basis for substantial equivalence (SE).”
Previously, this program was limited to review of device changes that did not affect the intended use or fundamental scientific technology. The Special 510(k) Program will focus on how the changes were evaluated, and a summary analysis of risk associated with the changes. This program will rely heavily upon the manufacturer’s design control and risk management processes, with a specific emphasis on design verification and validation activities.
This program will generally not be appropriate for “devices that manufacture a biological product at the point of care” because there would likely be no “well-established method to evaluate changes” and/or provide a summary analysis of risk.
The guidance document provides a useful 510(k) flowchart (Fig. 1) to aid the practitioner in determining if the change is eligible for the program. The Guidance also provides three very useful appendixes: Appendix A) Recommended content of a Special 510(k), Appendix B) Examples of changes, and Appendix C) Examples of the summary of design control activities that provide additional information to aid in the special 510(k) regulatory submission process.
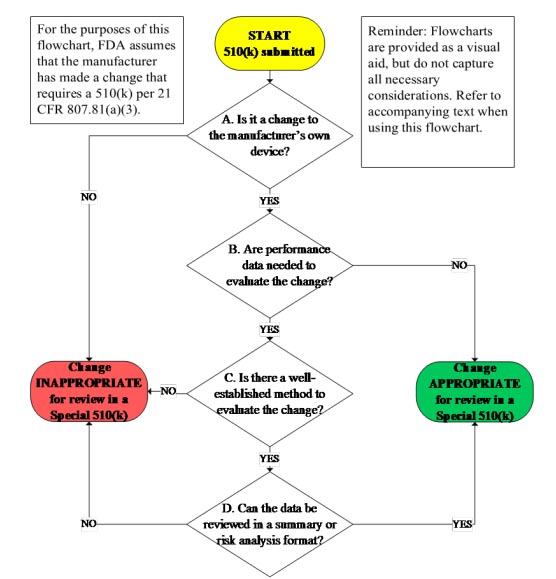
Fig. 1 — Special 510(k) flowchart. Image courtesy of the FDA.
The Abbreviated 510(k) Program
The Abbreviated 510(k) Program, published Sept. 13, 2019, supersedes the Abbreviated 510(k) content from The New 510(k) Paradigm - Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications, issued March 20, 1998. The Abbreviated 510(k) Program was intended to create an efficient submission preparation and review process that relies on guidance documents, special controls, and/or voluntary consensus standards.
The program will leverage testing recommended in guidance documents, special controls, or voluntary consensus standards and the resulting summary reports. For a regulatory submission that relies on guidance documents, special controls, or voluntary consensus standards, the summary report should describe how the guidance documents were used. When guidance document recommendations are not are fully utilized by the manufacturer, any deviation or alternative method used to demonstrate substantial equivalence must be justified in the regulatory submission.
Format for Traditional and Abbreviated 510(k)s
Format for Traditional and Abbreviated 510(k)s, published Sept. 13, 2019, supersedes Format for Traditional and Abbreviated 510(k)s - Guidance for Industry and FDA Staff, issued Nov. 17, 2005. This guidance provides information on how to properly format a regulatory submission for a Traditional or Abbreviated premarket notification 510(k).
This guidance does not apply to Special 510(k)s, premarket approval applications (PMAs), Humanitarian Device Exemption (HDE) applications, De Novo requests, or investigational device exemption (IDE) applications.
The FDA recommends a Traditional or Abbreviated 510(k) submission include all 20 section headings as described in the guidance. Any heading that does not apply should be marked as “This section does not apply” or “N/A” under that heading. Appendix A of the guidance, 510(k) Cover Letter, provides the recommended information to be included in the cover letter to aid the reviewer.
Refuse to Accept Policy for 510(k)s
Refuse to Accept Policy for 510(k)s, published Sept. 13, 2019, supersedes Refuse to Accept Policy for 510(k)s, issued Feb. 21, 2019. The purpose of this policy is to assess “whether a premarket notification (510(k)) submission meets a minimum threshold of acceptability and should be accepted for substantive review.”
It is important to understand this policy does not determine regulatory approval for the submission; this administrative review is used to determine the “completeness” or “readiness” of the application for further substantive technical review.
The guidance provides three useful checklists that should be used to assess completeness of the submission materials:
- Appendix A — Acceptance Checklist for Traditional 510(k)s
- Appendix B — Acceptance Checklist for Abbreviated 510(k)s
- Appendix C — Acceptance Checklist for Special 510(k)s
The FDA recommends the manufacturer should include the appropriate completed checklist as part of the submission package.
Conclusion
Recent changes in organization, structure, and philosophy at the FDA are a positive sign for the medical device industry. These new guidances are designed to make the regulatory submission process more efficient for the FDA and manufacturers. These programs are consistent with the “FDA’s statutory mission to protect and promote human health and FDA’s commitment to helping patients gain timely access to new medical devices that are high quality, safe and effective by using efficient review practices consistent with least burdensome principles.”
About the Author
 Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is Managing Principal Consultant at Quality Systems Compliance LLC, an ASQ Fellow and SRE Fellow. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications including; CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him with any questions or comments.
Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is Managing Principal Consultant at Quality Systems Compliance LLC, an ASQ Fellow and SRE Fellow. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications including; CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him with any questions or comments.