
Losing Sight Of The Intended Purpose Of Specifications Has Consequences For The CMO-Sponsor Relationship
By Julia O’Neill, Principal, Tunnell Consulting
 If you’re reading this article, you probably spend most of your working hours focused on creating a reliable supply of medicines at a reasonable cost. But most of us also have been customers for the products we manufacture. When we reach for a bottle of tablets or brace ourselves for an injection at the doctor’s office, we depend on that medicine being safe and effective in treating or preventing disease.
If you’re reading this article, you probably spend most of your working hours focused on creating a reliable supply of medicines at a reasonable cost. But most of us also have been customers for the products we manufacture. When we reach for a bottle of tablets or brace ourselves for an injection at the doctor’s office, we depend on that medicine being safe and effective in treating or preventing disease.
What matters to us as consumers is that the product embodies all the characteristics needed to treat or prevent our condition at the time we receive it. It’s an extremely rare patient who can describe in detail those characteristics. We take for granted that someone behind the scenes has figured out those requirements.
Sponsors of a new medicine submit an application defining critical quality attributes and their acceptable ranges. Health authorities review the application and then negotiate those attributes and limits with the sponsor. The resulting quality profile should provide us (consumers) assurance that the medicine is acceptable for its intended use. This fundamental purpose of specifications is a cornerstone of the ICH Guidances for Specifications for both chemical and biotechnological/ biological products.1, 2
HOW HAVE SPECIFICATIONS FOR PHARMACEUTICAL PRODUCTS DRIFTED FROM THEIR ORIGINAL PURPOSE?
In other industries, specifications are documented requirements agreed to by customer and supplier, typically without involving regulators in the negotiation. The preferred approach to setting specifications is to base them on required property values based on knowledge of the product’s use.3 These specifications are knowledge-based.
A very simple example is setting a mechanical tolerance for the dimensions of one part based on the dimensions of another part when those two parts must fit together to function properly. The dimensions can usually be measured with minimal variability. Specifications for jet fuel are another example of well-understood requirements.
When the true requirements aren’t fully known or can’t be determined without substantial measurement variability, specifications must be established on some other basis. Typical practice is to set specifications based on experience rather than knowledge by using the supplier’s demonstrated process performance or the quality level the customer is willing to accept.
A new procedure for establishing specifications for impurities acknowledges that impurity acceptance criteria often cannot be established by one definitive approach, but rather endorses clinical relevance as the guiding principle for developing specifications.4
Experience-based specifications miss the point. They are not based on knowledge of the product’s acceptability for its intended use, but instead reflect what’s been produced or consumed so far.
Current practices for specification-setting in the pharmaceutical industry have drifted from the intended purpose of specifications. There is growing recognition of this disconnect, resulting in a number of industry teams, regulatory initiatives, and conference sessions on “Clinically Relevant Specifications” — a more elegant term describing knowledge-based specifications for medicines. These align with many patient-focused initiatives within the FDA.5
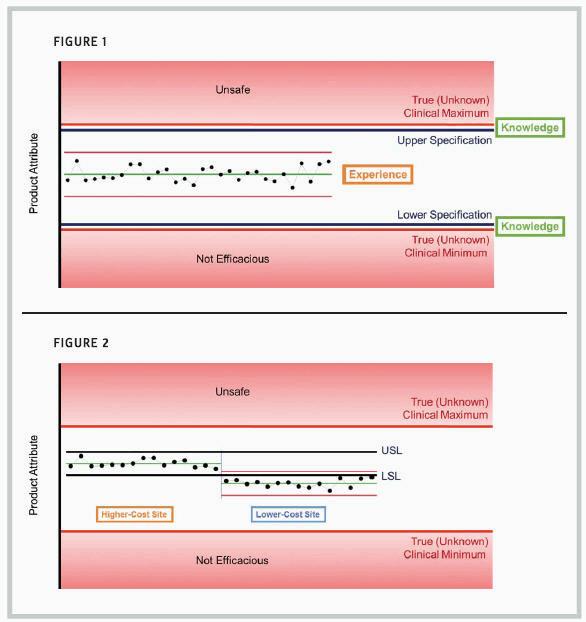
The scenario for setting clinically relevant specifi cations is illustrated in figure 1. This hypothetical product attribute would become unsafe for patients above its true clinical maximum. The same attribute would not be sufficiently effective below its true clinical minimum. The true minimum and maximum are typically not known exactly, and the opportunity to know those levels depends on deep knowledge of the product’s function in patients.
Ideally, clinically relevant specifications would be set just inside the range of the true clinical limits. These limits are based on knowledge of the product’s true requirements for patients.
Within the specification range, statistical process control (SPC) methods would be routinely applied to monitor process performance and support continuous improvement.6 These limits are based on manufacturing experience. In the pharmaceutical industry, confusion between statistical control limits and specifications impedes progress toward continuous improvement and cost reduction.
WHAT HAPPENS WHEN SPECIFICATIONS ARE BASED ON EXPERIENCE?
Current practices for setting specifications rely on process or clinical experience rather than knowledge of the product’s function in patients. Typically, the sponsor proposes specification limits based on the history of product attributes for the initial manufactured lots. These are sometimes referred to as “process capability limits.”
This approach requires lengthy process experience — 30 lots at a minimum — to provide a reliable forecast of future manufacturing performance. Statistical calculations such as tolerance intervals are applied to the initial manufacturing data to establish limits that will include most future results, assuming the process remains in control. (Other statistical approaches, such as the mean ±3 standard deviations or the range from minimum to maximum, are also sometimes applied, but I do not recommend these.)
Specifications based on initial process experience have a number of serious impacts, including potential disruption to outsourcing relationships. These consequences include:
Initial manufacturers with poorer control are “rewarded” with wider specifications, providing greater flexibility for manufacturing. Manufacturers with tighter control initially are “punished” with tight, less-flexible specifications.
Quality metrics such as process capability lose all meaning.
Specifications based on the initial manufacturing site limit flexibility in the supply chain.
Opportunities to reduce manufacturing cost or otherwise make continuous improvements are impeded by unnecessary regulatory barriers to change.
THE IMPACT ON CMO-SPONSOR RELATIONSHIPS
Let’s explore the damage experience-based specifications can inflict on the CMO-sponsor relationship. Using our example of a hypothetical product attribute, suppose specifications are set based on 30 manufactured lots produced at the initial manufacturing site. The left side of figure 2 shows 15 of those initial 30 lots, with specifiations labeled “USL” (upper specification limit) and “LSL” (lower specification limit).
That initial manufacturing site may carry somewhat higher costs than an alternative CMO site that is fully capable of making the same product. Suppose the CMO site is engaged to begin manufacturing the product at a lower cost for the sponsor. The results for the first 15 lots from the lower-cost CMO are shown to the right of the initial 15 lots.
The specifications were set artificially tight based on experience at the initial site. These specifications are disconnected from the true clinical requirements.
The CMO will be challenged to provide root cause for the difference in results or to “correct” its process to establish comparability between the two sites. All of the lots manufactured at the initial and CMO sites would be fully acceptable for patient use, but without knowledge of the true clinical limits, a great deal of effort will be expended to reconcile the difference.
It’s worth noting that the same difference would create non-value-added work even if the change in product attribute resulted from a continuous improvement made at the original manufacturing site. But the CMO-sponsor arrangement adds a layer or two of complexity to this already difficult situation.
WHAT WOULD IT TAKE TO RESTORE SPECIFICATIONS TO THEIR INTENDED ROLE?
Like so many other long-standing problems, if it was easy to correct this situation, it probably would have been corrected a long time ago. But our changing environment provides new opportunities to make progress toward clinically relevant specifications.
The best possible solution is to develop deep clinical knowledge of the action of pharmaceutical or biotechnological products in treating or preventing disease. The revolution in understanding of disease, and particularly its relation to genetic factors, puts this knowledge tantalizingly within reach. True clinically relevant specifications may emerge within the next few years, as clinical science advances for breakthrough products emerging from development pipelines. Investments in translational medicine support this trend.
The call to action for manufacturers is to embrace collaboration with clinical functions. In many large organizations today, clinical experts and CMC (chemistry, manufacturing, and controls) experts may have little interaction. Organizational barriers will need to be knocked down to turn scientific knowledge of true clinical requirements into manufacturing specifications.
Another accelerating factor could be the explosion of Big Data applied to patient experience. The connection of clinical outcomes to product properties was prohibitively labor-intensive just a few years ago. But advances in data systems and analytics are paving the way for data- based approaches to build deep knowledge of clinical performance in relation to product attributes.
The adoption of clinically relevant specifications as a standard for pharmaceutical manufacturing supports the simultaneous attainment of two worthy goals: providing a reliable supply of safe and efficacious products to patients, while reducing manufacturing costs.
1 ICH (1999) Q6A Specifi cations: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Product: Chemical Substance.
2 ICH (1999) Q6B Specifi cations: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products.
3 ASQC Chemical & Process Industries Division (1996) Specifications for the Chemical & Process Industries.
4 OPQ MAPP (2018) Establishing Impurity Acceptance Criteria As Part of Specifications for NDAs, ANDAs, and BLAs Based on Clinical Relevance.
5 OPQ White Paper (2015) FDA Pharmaceutical Quality Oversight: One Quality Voice. https://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM442666.pdf
6 Montgomery, Douglas C. (2013) Introduction to Statistical Quality Control, 7th edition.