
The Global Regulatory And Quality Environment For Biopharma Outsourcing
By Bikash Chatterjee
 The topic of rising healthcare costs isn’t just a first-world issue anymore.
The topic of rising healthcare costs isn’t just a first-world issue anymore.
Global healthcare expenditures are rising, and spending is increasing at an annual rate of 5.4 percent between 2017-2022, from $7.724 trillion to $10.059 trillion, according to Deloitte’s 2019 Global Healthcare Outlook. The global drug market will continue to grow, driven in part by double-digit economic growth in India and China and by downward pricing pressures in the U.S. The new regulatory frameworks now deployed in China are fueling growth in Asia Pacific.
While the U.S. and Western Europe still make up more than half of the global market, China has replaced Western Europe as the second-largest marketplace. Harmonization of standards is inevitable as the socioeconomic statuses of these markets converge. Therefore, it is critical that organizations looking to engage an external contract service provider be aware of these newly established regulations to align their programs with the latest expectations for each relevant market. Let’s examine these in detail.
EUROPEAN UNION
The EU is undergoing major changes in the pharma, clinical data, and medical device arenas (Figure 1).
Several new regulations are worth noting:
Identification of Medicinal Products (IDMP): Data Standards
The European Medicines Agency (EMA) is implementing the ISO IDMP standards for the identification of medicinal products in a phased program, based on the four domains of master data in pharmaceutical regulatory processes: substance, product, organization, and referential (SPOR) data. Under the IDMP standards, pharmaceutical companies will be required to electronically submit detailed product data and maintain it on an ongoing basis.
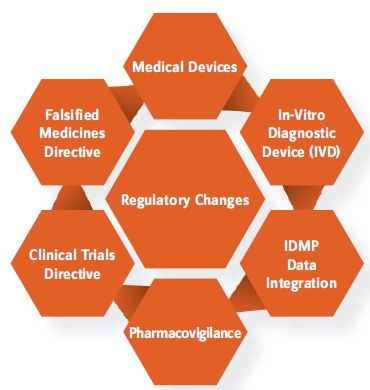
Figure 1: EU Regulatory Changes
The goals of this new standard are to:
- help facilitate the creation of global drug dictionaries and product dossiers
- link product and safety information across global regulatory agencies
- increase the industry’s signal detection capabilities to quickly identify product risks and issues, including coordinating product recalls
- connect critical product information within healthcare systems.
The new framework consists of five ISO standards, shown in Figure 2. Becoming IDMP-compliant will drive pharmaceutical companies and full-service contract service providers to make significant changes to current product-related processes and systems, in a new era of cross-functional collaboration that paves the way for transformational benefits that extend beyond compliance.
CMOS NEED TO UNDERSTAND:
- evolving regulations, implementation guidelines, and iterations
- the compliance timeline and consequences of not meeting regulations
- the IDMP data model and where data resides in the organization.
Medical Device Regulation (MDR)
In June 2016, the European Parliament and the Council of the European Union adopted the far-reaching EU Medical Device Regulation following calls for greater control and stringent monitoring of medical devices, triggered by the Poly Implant Prothèse (PIP) breast implant scandal, a widespread hip replacement recall, and other incidents that revealed the system’s regulatory weaknesses. This regulation goes into effect in May 2020 and will transform both the medical device classification and the approval process. The MDR regulation will supersede all prior device approvals within the EU, with no grandfather clause for the former regulation. Compliance for reclassified devices must be in place by May 2020, or the product must be withdrawn from the market.
Key changes within this new regulation involve:
- Scrutiny process: The European Commission (EC) will be able to review recommendations for Conformité Européenne (French) (CE) marking prior to approval.
- Common technical specifications (CTS): The EC’s ability to create common technical specifications will be expanded to all devices.
- New rules for notified bodies: Only newly created special notified bodies will be able to issue CE certificates for high-risk devices such as implants.
- Audits for notified bodies: Notified bodies will be audited for compliance with the new regulations jointly by two competent authorities (i.e., the regulatory body for each member state). Also, manufacturers will be subject to unannounced audits by notified bodies.
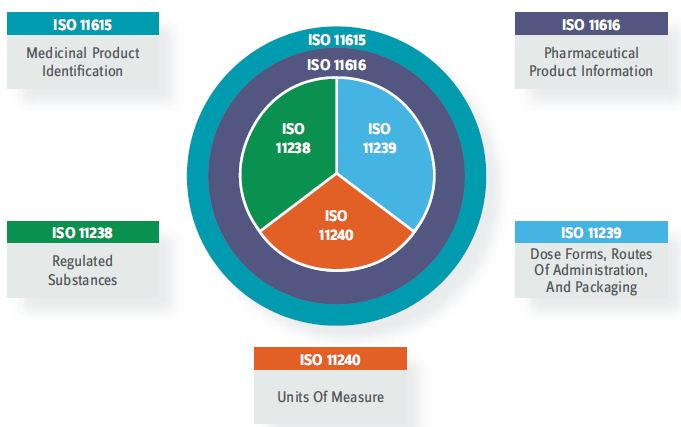
Figure 2: IDMP ISO Standards
- Reclassification of medical devices: Spinal implants, devices that control and monitor active implants, nanomaterials, apheresis machines, and combination products will be reclassified as Class III devices requiring design dossiers.
- Identification and traceability of devices: A unique device identification (UDI) system will be required for labeling, and the European Databank on Medical Devices (EUDAMED) will be expanded. Manufacturers will need to provide a summary of safety and clinical performance for Class III devices and also for implants of lower classification.
- Clinical evaluation and investigations: The new MDR regulation will put in place a regimen for clinical investigations with a mandatory post-market and clinical follow-up (PMCF) and periodic safety update reports.
- Post-market surveillance (PMS), vigilance, and market surveillance: Under the regulation, PMS and vigilance requirements will be revisited, and manufacturers will consequently need to amend their procedures.
- Change in format of technical files: Formatting declarations of conformity and technical files is revised under the new regulation. This requires manufacturers to create a summary document for each section instead of providing complete protocols and reports.
CHINA NMPA
China’s regulatory framework is moving into close alignment with global regulatory practice, and few regulatory bodies have encountered as much change in a short period of time as China’s National Medical Products Administration (NMPA).
As ICH guidelines become China’s standard, China is increasingly willing to accept global clinical data in support of local product registrations, with priority for products that serve Chinese patients’ unmet medical needs.
China has implemented several key changes to accelerate clinical trial and drug approval timelines.
- Inclusion of data from clinical trials undertaken outside China. Drug sponsors and CROs that are attentive to the NMPA’s requirements will be well positioned for access to the Chinese market.
- Streamlined clinical trial approvals (CTAs). Specifically, the NMPA is allowing clinical trial materials to be tested by the sponsor or a trusted third-party testing lab, rather than having to be tested by a government-accredited testing lab.
- Lifting of restrictions on the involvement of Chinese sites in multicenter Phase 1 studies. This changes the dynamic when selecting a CMO or CRO for multicenter Phase 1 studies.
- Fast-track approval for drugs and devices. Specifically, new drugs and devices in development that meet urgent clinical needs in China can be approved for marketing conditions if data from early- or mid-stage trials show promising clinical value. Further, new drugs or devices for rare diseases can be approved for marketing in China if they have been approved for marketing overseas.
What’s more, China’s revised Drug Administration Law (DAL) entered into effect in December 2019. Under the new DAL, the market authorization holder (MAH) system applies equally to imported and domestic drugs, with MAH responsibility for the entire life cycle of a drug. Marketing authorizations can be transferred from one company to another without changing contract manufacturers, subject to NMPA approval. The amended regulation will enable Chinese MAHs to work with overseas CMOs. Likewise, foreign MAHs may choose to work with CMOs in China and restructure their supply chains accordingly.
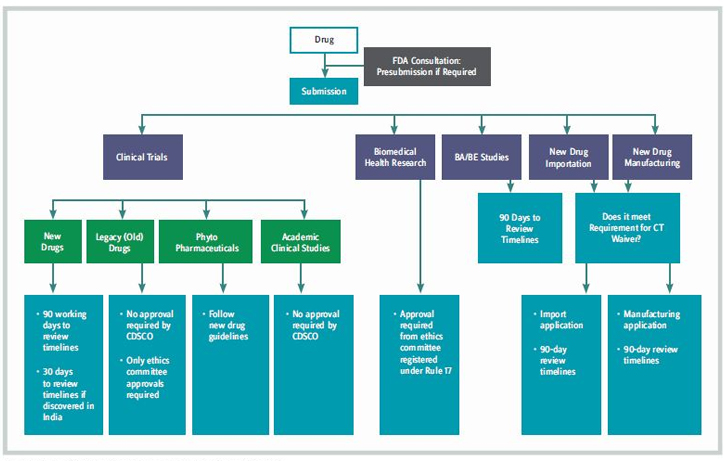
Figure 3: India’s New Framework For Product Registration
INDIA
The Indian health ministry announced that certain drugs approved for use in major markets (such as the EU and U.S.) will be automatically approved in India without a further native clinical trial having to take place, to give patients faster access to new medicines. The Ministry of Health & Family Welfare (MHFW) announced the new Drugs and Clinical Trials Rules 2019 in March 2019, to improve the ethical and quality standards of clinical trials in India. New guidance consists of 13 chapters (including 107 rules) and eight schedules that apply to all new drugs, as well as investigational new drugs for human use, clinical trials, bioequivalence and bioavailability studies, and ethics committees. The new framework for product registration is shown in Figure 3.
The new clinical trial rules include :
- approval for clinical trials in 30 working days for indigenous drugs to speed up the trial process and encourage local drug development
- provision for accelerated product approval, with some conditions, adding pre- and post-submission meetings with authorities to increase regulatory engagement.
The new framework is designed to stimulate the local clinical research industry, allowing more global clinical studies in India and promoting Indian indigenous drug development. These comprehensive new rules should improve the ethical and quality standards of clinical trials in India, aiding patients and industry.
The one constant we can count on is change. It has taken time, but the regulatory philosophies of the major markets are converging, creating avenues that accelerate access to new drug therapies while providing a solid, structured framework for clinical trial and regulatory oversight. Drug sponsors pursuing an outsourcing strategy will have to make sure the necessary processes and systems are in place — both internally and with their contract service providers — to ensure compliance in a new decade of modernized market regulatory expectations.
BIKASH CHATTERJEE is president and chief science officer for Pharmatech Associates. He has over 30 years’ experience in the design and development of pharmaceutical, biotech, medical device, and IVD products.