
The Importance Of Pursuing Clinical Trial Costs
By Amit Rathore
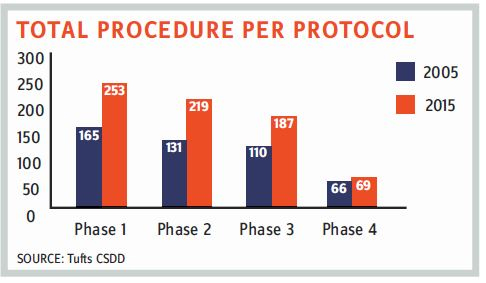
 There’s no doubt that clinical trials are becoming more complex in terms of regulatory compliance and scope of study. The FDA is stressing more on data integrity, and in December 2018, it finalized its draft of “Data Integrity and Compliance with cGMP Guidance.” The number of drug GMP warning letters has continuously increased; the FDA issued almost 200 percent more warning letters in 2018 as compared to 2015. Also, there is an increase in the number of total procedures per protocol involved in a typical clinical trial. Apart from increasing operational complexities, consolidation within the CRO industry in recent years has forced sponsor companies to be more vigilant and thorough when planning and overseeing outsourced clinical trials. Consequently, it is more important than ever that procurement teams develop a clear understanding of the various cost drivers related to operating a clinical trial.
There’s no doubt that clinical trials are becoming more complex in terms of regulatory compliance and scope of study. The FDA is stressing more on data integrity, and in December 2018, it finalized its draft of “Data Integrity and Compliance with cGMP Guidance.” The number of drug GMP warning letters has continuously increased; the FDA issued almost 200 percent more warning letters in 2018 as compared to 2015. Also, there is an increase in the number of total procedures per protocol involved in a typical clinical trial. Apart from increasing operational complexities, consolidation within the CRO industry in recent years has forced sponsor companies to be more vigilant and thorough when planning and overseeing outsourced clinical trials. Consequently, it is more important than ever that procurement teams develop a clear understanding of the various cost drivers related to operating a clinical trial.
CLINICAL TRIAL COST DRIVERS
Although there are numerous factors that affect the cost of a clinical trial, some of the major ones include:
- Increased costs of clinical supplies and equipment:The cost of clinical supplies is affected by many factors, such as manufacturer and packager audits, SOP training, and raw material and shipment costs.
- Extended timelines of clinical trials: The inclusion of more procedures and protocols has increased clinical trial timelines.
- Increased regulations, particularly at the clinical and CMC (chemistry, manufacturing, and controls) levels: During postdrug approval and commercialization, any change to the registered content as mentioned in record by the manufacturer must be conveyed to the regulatory authorities. These changes are categorized based on the nature of their complexities and must be approved by the regulatory authority. The International Council for Harmonization (ICH) Q12 guideline provides a framework for managing post-approval CMC changes for new and marketed pharmaceutical drug substances (APIs), drug products (DPs), and drug-device combination products. In mid-2018, the FDA published the ICH Q12 draft guidance on “Technical and Regulatory Considerations for Pharmaceutical Lifecycle Management.”
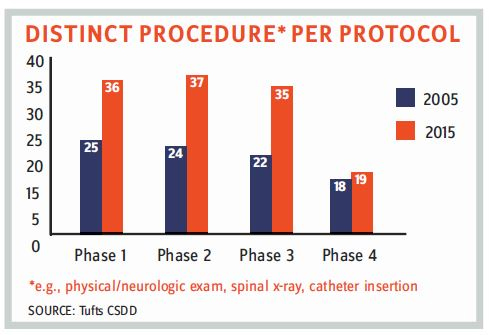
- Monitoring complexities: In addition to the increase in the number of clinical trial protocols and procedures, monitoring complexities also have increased. Global landscapes, lack of common formats, changing regulatory frameworks, and other variables increase the complexities for pharmaceutical companies. Primarily, lack of robustness in an organization’s change-control system leads to the possibility of regulatory noncompliance. According to the 2017 BIMO (Bioresearch Monitoring) program released by the FDA, the common deficiencies in sponsors, CROs, and monitors identified in FDA Form 483 are:
- inadequate monitoring
- failure to bring investigators into compliance
- inadequate accountability for the investigational product
- failure to obtain FDA and/or IRB (institutional review board) approval prior to study initiation.
- Patient-recruitment intricacies: Patient recruitment and retention is challenging for every clinical trial, primarily because of:
- lack of prospect of improved treatment
- inconvenience of sites
- high cost of medication
- frequent site visits
- long trial duration
- time and cost of travel
- fewer monetary or nonmonetary incentives
- social and emotional impact on patients, families, and caregivers.
- Workforce competence: According to the 2017 BIMO matrix, the common investigator deficiencies identified in FDA Form 483 are:
- failure to follow the investigational plan/agreement or regulations, or both — study subjects not meeting the protocol inclusion and exclusion criteria before enrollment
- protocol deviations
- inadequate recordkeeping — not maintaining drug disposition records
- inadequate subject protection — informed consent issues, failure to report AEs (adverse events)
- inadequate accountability for the investigational product
- inadequate communication with the IRB
- investigational product represented as safe/ effective.
- Data collection and synergy complexities: The global interests of pharmaceutical companies have spread the scope of clinical trials across the globe. Though the major regulatory guidelines are the same across the world, the practical incorporation and interpretation could differ regionally. Industry consolidation and adoption of new data-capture systems increase the difficulty in establishing data synergies and integrity.
Regulatory agencies are also pushing new regulations regarding data reporting. In March 2019, Health Canada announced the revision of the validation rules for “Regulatory Transactions in electronic Common Technical Document (eCTD)” format.
The USFDA also is pushing for the adoption of the eCTD format and has extended the compliance date for Type III Drug Master Files (DMFs), to be submitted in eCTD format by May 5, 2020.
CLINICAL TRIAL COST COMPONENTS
The cost of a clinical trial can be calculated by analyzing the cost incurred for each patient at each site during the study. To estimate the overall cost, many parameters must be considered. The following table captures a sample of the various cost components involved in a clinical trial.

Procurement teams must adopt a detailed approach to estimating the cost per component. Involvement of stakeholders from all departments is vital due to the complex regulatory environment; you could easily miscalculate a cost parameter if the correct operational teams are not consulted. For example, estimating the cost of data management is very tricky. Currently, regulatory authorities are focused on data integrity. Each data point could be influenced by the following factors:
- The patient recruitment process often requires patients to complete four or more questionnaires. This may or may not lead to any meaningful interpretation, but it does elongate the patient-recruitment process.
- During clinical trials, the costs of tests such as MRIs is higher (generally three to four times) than the cost of comparable tests during general routine medical visits. This is because universities and research organizations offset the grant money for the routine tests, or the government subsidizes these tests in hospitals for the general public.
- Sometimes, detailed biomarker analysis samples are sent to different external labs, attracting higher fees.
- Similarly, due to specific procedural requirements, it is possible that physicians perform specialized tests for particular disease indications along with regular physical examinations of patients. This may require more physician time, which results in higher costs.
- All the data collected is required to go through stringent data-management protocols to ensure data integrity and patient safety. This might require study coordinators to spend more time to fulfill the requirements of Good Clinical Practice guidelines, which further adds to the costs.
CLINICAL TRIAL COST DISTRIBUTION BY PHASE
According to the study “Estimated Costs of Pivotal Trials for Novel Therapeutic Agents Approved by the U.S. Food and Drug Administration, 2015-2016” published in JAMA Internal Medicine in 2018, the estimated costs of trials ranged from $2.1 million for a trial that enrolled four patients to test uridine triacetate for a rare hereditary metabolic disorder to $346.8 million for a noninferiority trial that assessed the impact of a new combination cardiovascular drug for chronic heart failure on hospitalization and cardiovascular mortality. The clinical trials cost a median of $41,117 per patient and $3,562 per patient visit. Clinical trial cost varies with the therapeutic area as well.
Such data points can help in estimating and benchmarking clinical trial costs. But a cautious approach must be adopted, as these costs could vary according to the particular requirement of drug/therapy/procedure. For example, clinical trial requirements of cell therapies are more stringent and cost-intensive than those of common drug therapies.
AMIT RATHORE is principal analyst at Beroe Inc. and an Institute for Supply Management (ISM) Certified Professional in Supply Management (CPSM).